
iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建(全文翻译/PPT/视频解读)
- 看不见的线
- 5410
- 2024-06-04 09:04:44
- 文章来源:iMeta

干旱生态系统中土壤真菌与细菌群落构建的关系
Linking soil fungi to bacterial community assembly in arid ecosystems
DOI:https://doi.org/10.1002/imt2.2
发表日期:2022年2月24日
第一作者: Shuo Jiao(焦硕)1
通讯作者:Weimin Chen(陈卫民)(chenwm029@nwsuaf.edu.cn)1, Gehong Wei(韦革宏)(weigehong@nwafu.edu.cn)1
合作作者: Haiyan Chu(褚海燕), Baogang Zhang(张宝刚), Xiaorong Wei(魏孝荣)
主要单位:
1西北农林科技大学(State Key Laboratory of Crop Stress Biology in Arid Areas, Shaanxi Key Laboratory of Agricultural and Environmental Microbiology, College of Life Sciences, Northwest A&F University, Yangling, China)
图型摘要
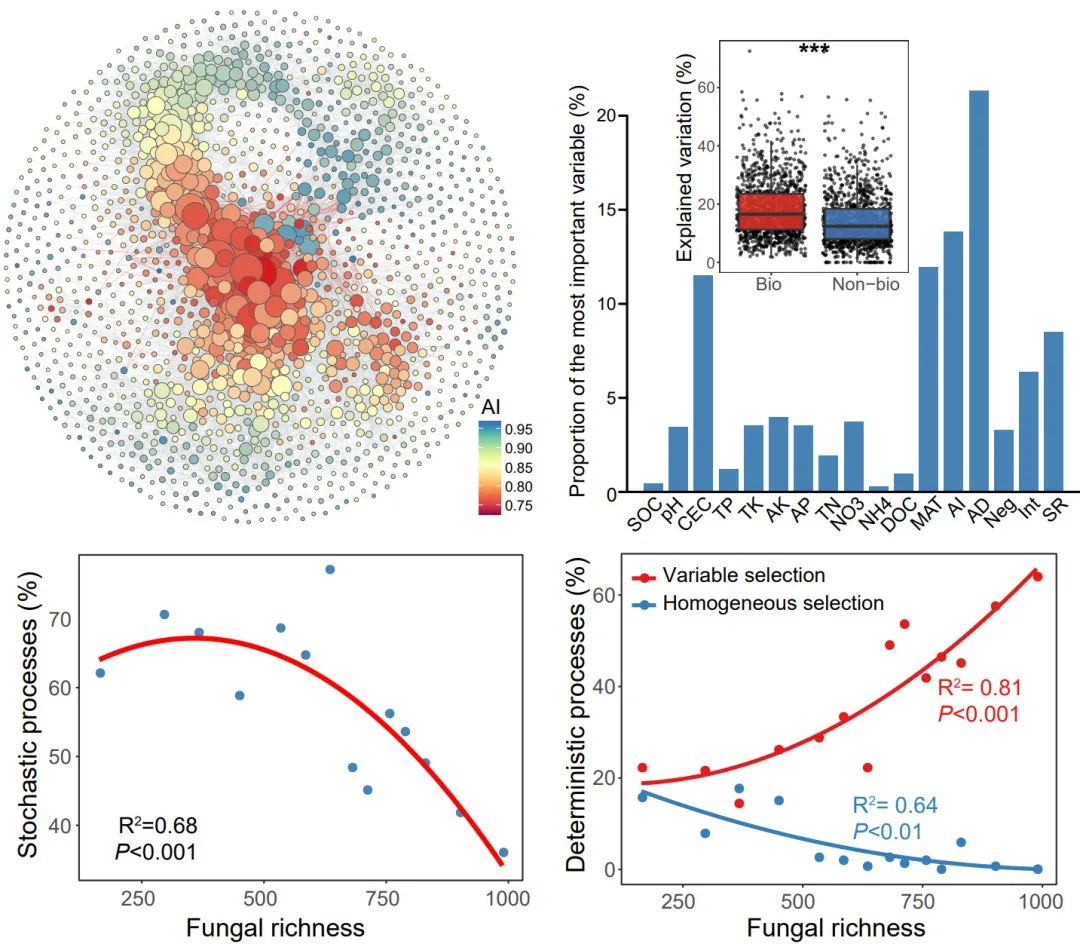
土壤真菌丰富度调节细菌群落的组成
随机过程随真菌丰富度的增加而减少
干旱的增强会降低细菌α-多样性
作者视频解读
Bilibili(有字幕):https://www.bilibili.com/video/BV1Sb4y1s7HD/
中文翻译、PPT、视频解读等扩展资料下载,请访问期刊官网:http://www.imeta.science/
摘要
阐明生物因素在驱动群落构建中的作用,对于理解多样性和生态系统功能至关重要,是微生物生态学中一个基本课题。本研究将跨生物群落的观测研究与微宇宙实验相结合,研究西北旱区复杂陆地生态系统中生物因素(如土壤真菌和跨界物种的相关关系)对土壤细菌生物地理和群落构建的影响。结果表明,土壤中真菌丰富度调节了细菌群落构建过程的平衡,随机过程随着真菌丰富度的增加而减少。进一步研究结果表明,由于气候变化导致的干旱增强,细菌α-多样性将减少,特别是在荒漠土壤和下层土壤,并且导致物种间更多的负相关性。该项研究有助于我们对干旱生态系统中土壤真菌与细菌的生物地理格局和群落构建机制之间联系的理解。
关键词: 群落构建,生物因素,干旱生态系统,生物地理学,细菌
引言
土壤微生物在陆地生态系统的各种生态过程中发挥着重要作用,包括土壤中的养分循环、污染物降解以及在环境变化下维持生态系统服务与功能的稳定。揭示微生物群落多样性和生物地理格局的基本机制,对于确定其与群落稳定性和生态系统功能之间的联系至关重要,是群落生态学研究的重要课题。微生物生物地理学的观点是,微生物无处不在,但受环境选择,突出了微生物很强的扩散能力和生态位适合度。然而,人们普遍认为确定性过程和随机过程都会影响微生物群落的生物地理格局和距离衰减关系(即微生物群落相似性随着地理距离的增加而减小)。确定性过程指非随机的基于生态位的机制,包括环境过滤和种间相互作用(如竞争、促进、互惠和捕食)。而随机过程主要反映物种相对丰度的随机变化,包括随机的出生、死亡和扩散事件。已有很多研究在不同生境和不同尺度(如区域、大陆和全球)量化了随机性过程和确定性过程驱动微生物群落构建的相对重要性,但依旧存在持续的争论。目前,研究复杂陆地生态系统中微生物群落空间变化的整个过程仍然是一个挑战。
在确定性和随机性过程的框架下,以往的研究主要集中在大尺度范围内非生物因素(如土壤、气候和地理)对微生物群落结构的影响。例如,土壤pH、养分、土壤质地、气候条件等环境因素对微生物群落地理分布的显著影响。然而,很少探究微生物类群之间的生物相互作用在塑造群落构建中的作用,然而生物相互作用可以决定微生物群落的功能属性或生态位占有。例如,在环境异质性下,由于适合度差异引起的物种间相互作用(如竞争和互惠)的变化会导致群落的生态位划分。这可以根据物种共现模式及其网络拓扑性质来推断。微生物 β 多样性无法被解释的部分可能与不同空间尺度微生物网络中物种的共发生模式和拓扑特征有关。
阐明调节随机过程与确定性过程之间平衡的因素有助于对群落构建机制的理解。例如,土壤pH值的变化可能会塑造土壤细菌群落的构建过程;农田生态系统中,硫对土壤真菌群落的随机和确定性过程起着调节作用,硫含量较高时,土壤真菌群落主要由随机性过程驱动。特别是真菌和细菌之间的相互作用在土壤中较为常见,在很多生态过程中发挥着重要作用。例如,土壤细菌和真菌共享资源,对基质的竞争可能引起细菌和真菌之间的拮抗作用;土壤真菌可能主要分解难降解有机物,如木质素,而细菌可能共生利用真菌衍生的基质。
本研究采用跨生物群落的观测数据和微宇宙实验探究生物因素对细菌群落构建的影响。首先对河西走廊(中国西北旱区典型绿洲-荒漠过渡带)251个土壤样品进行分析。探究土壤细菌群落在大尺度上的生物地理格局,量化了非生物因素和生物因素(土壤真菌和跨界物种关联)对细菌β-多样性和群落构建的相对贡献。本研究还利用微宇宙控制实验探究了真菌丰富度对土壤细菌群落结构的影响。在此,假设(1)微生物类群间的相互作用将导致了群落 β-多样性在大尺度上的显著变化;(2)土壤真菌介导土壤细菌群落构建过程的平衡。本研究揭示了生物因素在复杂陆地生态系统对细菌分布和群落构建的重要影响,这是土地利用变化情况下不可忽视的。
结果
河西走廊土壤细菌α多样性的分布格局
基于河西走廊251个跨生物群落土壤样品(样带间隔1257.6km),预测了土壤细菌α-多样性的空间分布(Shannon指数;图1a)。细菌在低纬度和高经度区域的α-多样性较高。大部分序列属于变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)和绿弯菌门(Chloroflexi)等,并且显示出不同的跨生物群系分布(图1B)。变形菌纲在湿地土壤中较为丰富,而酸杆菌纲的相对丰度在农业和森林土壤中显著较高。荒漠土壤放线菌的丰度较高。基于相关性的跨界共现网络,探究土壤微生物群落的共现模式,包括土壤细菌和真菌分类群。网络由1740个节点(即ASV)和7088条边组成(图1C)。通过计算每个分类单元所偏好的环境条件,发现类群对干旱(AI)的偏好与网络节点度显示出最高的相关性(Pearson’ r = 0.321, P < 0.001),随着AI的增加,节点度显著降低(图1D)。这表明在较低的AI环境中,微生物类群之间的相互联系更紧密,更有利于微生物共存。然后,基于单个土壤样本节点,提取的子网络计算其拓扑特征,以细菌和真菌类群间的平均程度(AD)、负相关的比例(Neg)和相互关联的比例(Int)作为生物因子。随机森林(图S2A)和多元回归(图S2B)分析表明,AI是预测上述三种拓扑特征最重要的变量。Neg与AI呈显著正相关,AD与Int呈负相关。
图1. 土壤细菌多样性与共现网络的一般模式
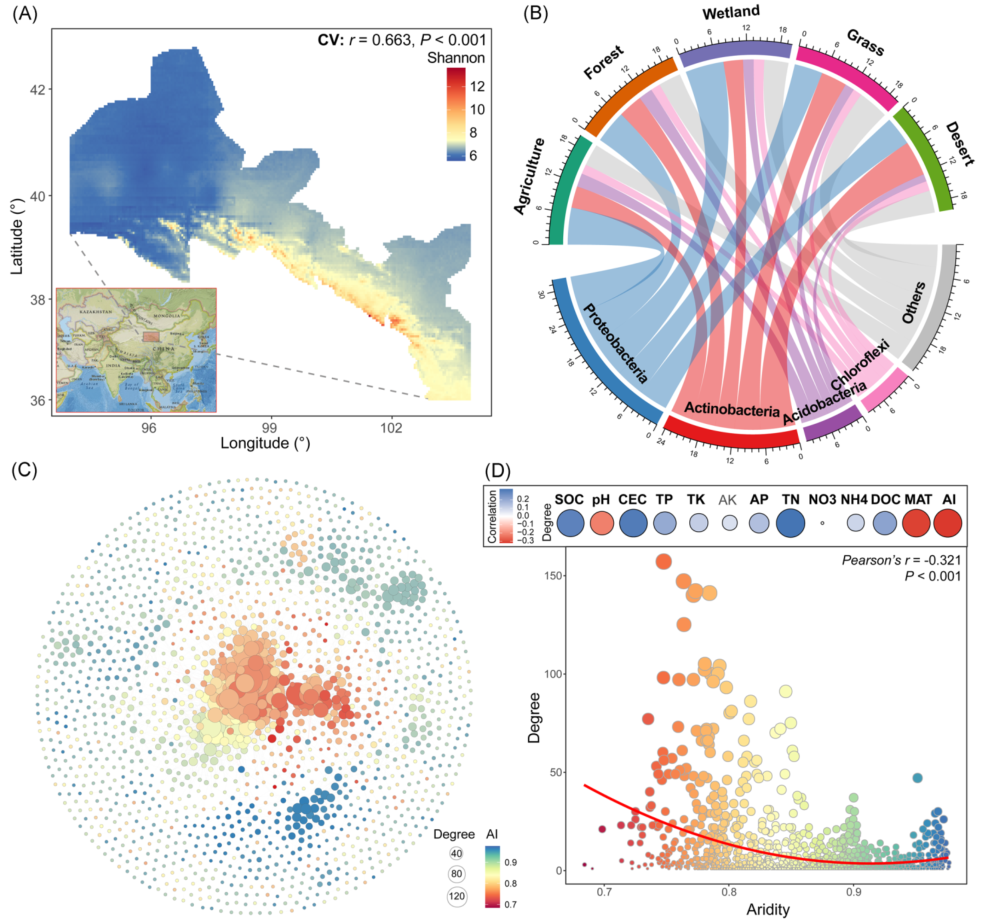
(A) 利用co-kriging插值方法预测细菌α-多样性的空间分布(Shannon指数);
(B) 在门水平上显示土壤细菌类群在不同生物群落中的分类学分布的弦图。每个条带的宽度代表不同门的细菌类群的相对丰度;
(C) 微生物类群跨界共现网络。网络的颜色是根据类群对干旱的偏好而定的。连接表明强烈而显著的(P < 0.001)相关性,分为正(Spearman’s ρ > 0.6; 深灰色)或负(Spearman’s ρ < -0.6; 红色)边。每个节点的大小与ASV的度成正比;两个节点(即一条边)之间的连接的宽度与斯皮尔曼相关系数的值成正比;
(D) 生态网络中微生物类群的度与其环境偏好之间的相关性(上图)。微生物类群度与干旱环境偏好的线性关系(下图)。
非生物(如土壤和气候)和生物因素中,真菌丰富度、AD和AI对解释不同生境细菌α-多样性变化的贡献最大(图2A)。AI与细菌α-多样性之间存在显著负相关,而细菌α-多样性随着真菌丰富度和AD的增加而增加(图2B)。AI对荒漠土壤细菌α-多样性的负影响最大,其次为农业土壤;而AI对森林土壤中细菌α-多样性的影响最弱(图2B)。此外,AI对各生境下细菌α-多样性的影响在下层比在上层更强(图2C),说明在不同生境下,AI对下层细菌α-多样性的影响更大。
图2. 不同生物群落土壤细菌α-多样性的驱动因素
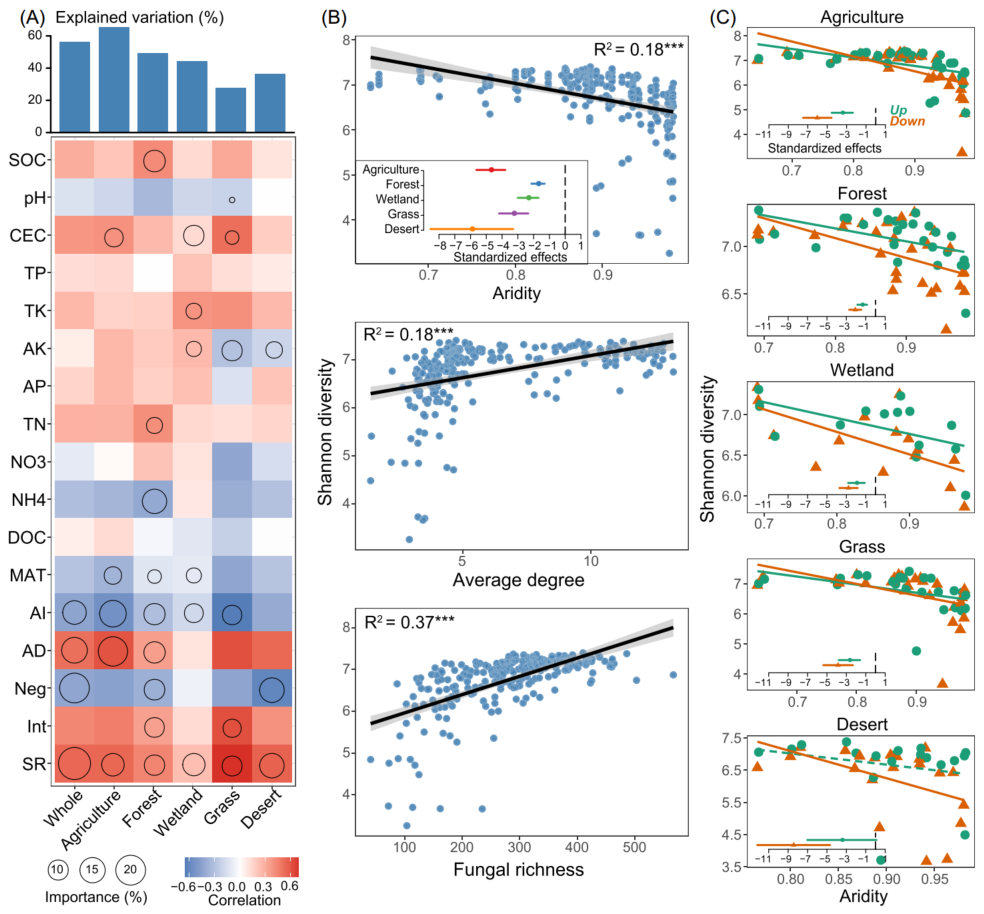
(A) 基于相关性和随机森林模型预测非生物和生物因素对细菌α-多样性的相对贡献。圆的大小表示变量的重要性(即随机森林模型计算的均方误差增加的百分比)。颜色表示斯皮尔曼相关性。AD,平均度;Neg,负相关的比例;Int,细菌和真菌之间相互作用的关联比例;SR,土壤真菌丰富度;
(B) 通过线性最小二乘回归分析,估计细菌α-多样性与主要驱动因子之间的关系。比较了不同生物群落中干旱对细菌α-多样性的标准化影响[标准化的斜率(mean ± s.e.m.)];
(C) 通过线性最小二乘回归分析,估算了干旱与表层(0-15 cm)和次表层(15-30 cm)细菌α-多样性的关系。比较了标准化干旱条件对两层细菌α-多样性的影响。
河西走廊土壤细菌β多样性格局
采用主坐标约束分析(Constrained analysis of principal coordinates, CAP)进一步探究非生物因素和生物因素对细菌β-多样性的影响。AI和AD对细菌β-多样性的影响最大(图3A)。通过Mantel和偏Mantel检验进行量化环境、地理和生物变量对细菌β-多样性变化的相对贡献。生物因子比环境和地理因子更能预测土壤细菌β-多样性,表明生物相互作用对土壤细菌β-多样性的驱动作用更强。为了检验生物因子是否在决定群落中特定成员的分布中发挥主要作用,结合多元回归模型和方差分解分析来量化生物因素对每个优势分类单元的贡献,这些优势分类单元指丰富 (相对丰富的前10%)且普遍存在 (在>50%的样本中存在)。结果表明,AD是最佳预测因子,其次是AI(图4)。此外,与只考虑非生物因素相比,当考虑生物因素时,观察到更多的可解释变异。表明生物因子在控制特定细菌类群的构建中起重要作用。
图3. 不同生物群落中细菌β-多样性的生物地理格局
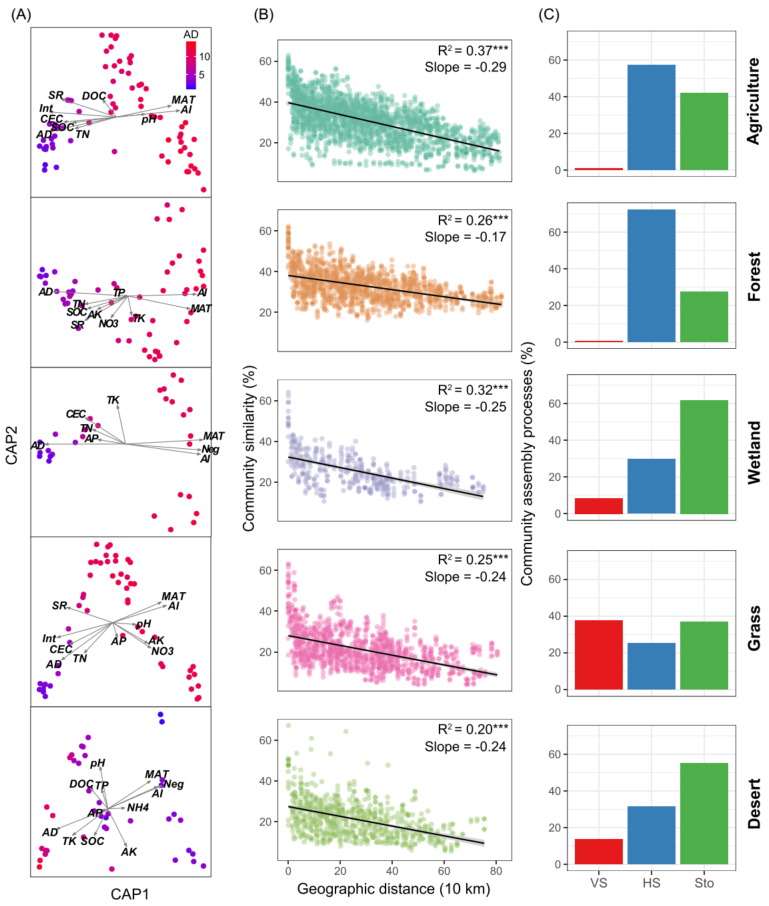
(A) 主坐标约束分析(CAP)显示影响细菌构建的非生物和生物因素。样本点按平均度(AD)着色;
(B) 显示Bray-Curtis相似性与采样点之间地理距离的距离衰减曲线。实线表示普通的最小二乘线性回归。星号表示显著相关(*, P < 0.001);
(C) 土壤细菌群落的周转率主要受确定性[同质选择(HS)和变量选择(VS)]和随机性过程(Sto)的控制。
图4. 优势细菌类群的系统发育分布及其主要驱动因素
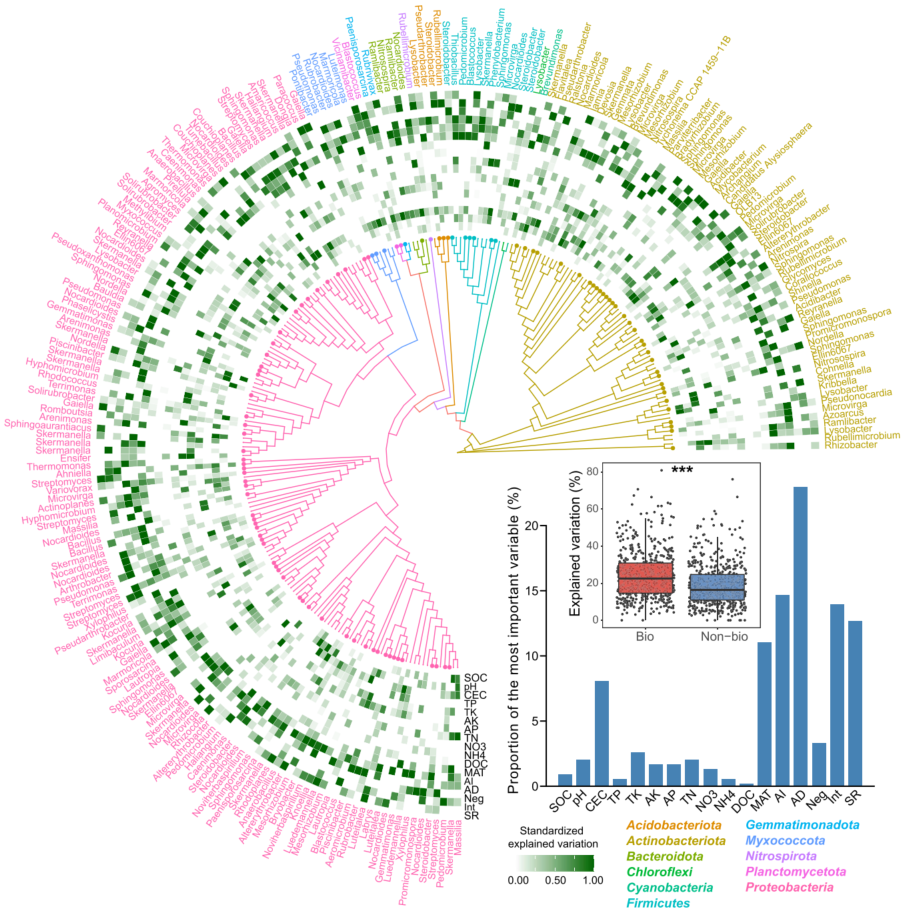
本分析中选择了高度丰富(相对丰度前10%)和普遍存在(在所有土壤样品中都存在)的ASVs。采用邻接法构建系统发育树。通过多元回归模型和方差分解分析,热图显示了非生物和生物因子在解释所选优势类群方面的相对重要性。条形图显示了解释主要分类单元丰度变化的最重要变量的比例。箱线图表明考虑生物和非生物因素(Bio)和只考虑非生物因素(Non-bio)之间的解释差异。
土壤细菌群落构建及其影响因素
细菌群落的相似性呈现出显著的地理衰减关系(DDR)(图3B)。然而,由线性回归模型估计的距离衰减斜率表明在不同的生境中有所不同。农业土壤的斜率(斜率= -0.37)显著高于其他生境,森林土壤的斜率(斜率= -0.17)最平坦。然后估计了驱动DDR的群落构建机制。零模型显示,在农业、森林和草地土壤中,细菌群落主要由确定性过程驱动,而在湿地和荒漠土壤中,细菌群落主要由随机过程驱动(图3C)。其中,农田和森林土壤中细菌的群落构建过程主要由同质选择主导,草地中细菌的群落构建过程主要受变量选择驱动。Mantel检验结果表明,真菌丰富度是βNTI的最佳预测因子,βNTI值与真菌丰富度差异的两两比较(图5A和5B)验证了此结果。随着真菌丰富度的增加,随机过程对细菌群落的影响逐渐减小,同质选择和变量选择的相对影响分别增大和减小(图5C和5D)。
图5. 土壤真菌丰富度对土壤细菌群落的确定性和随机性过程的相对影响
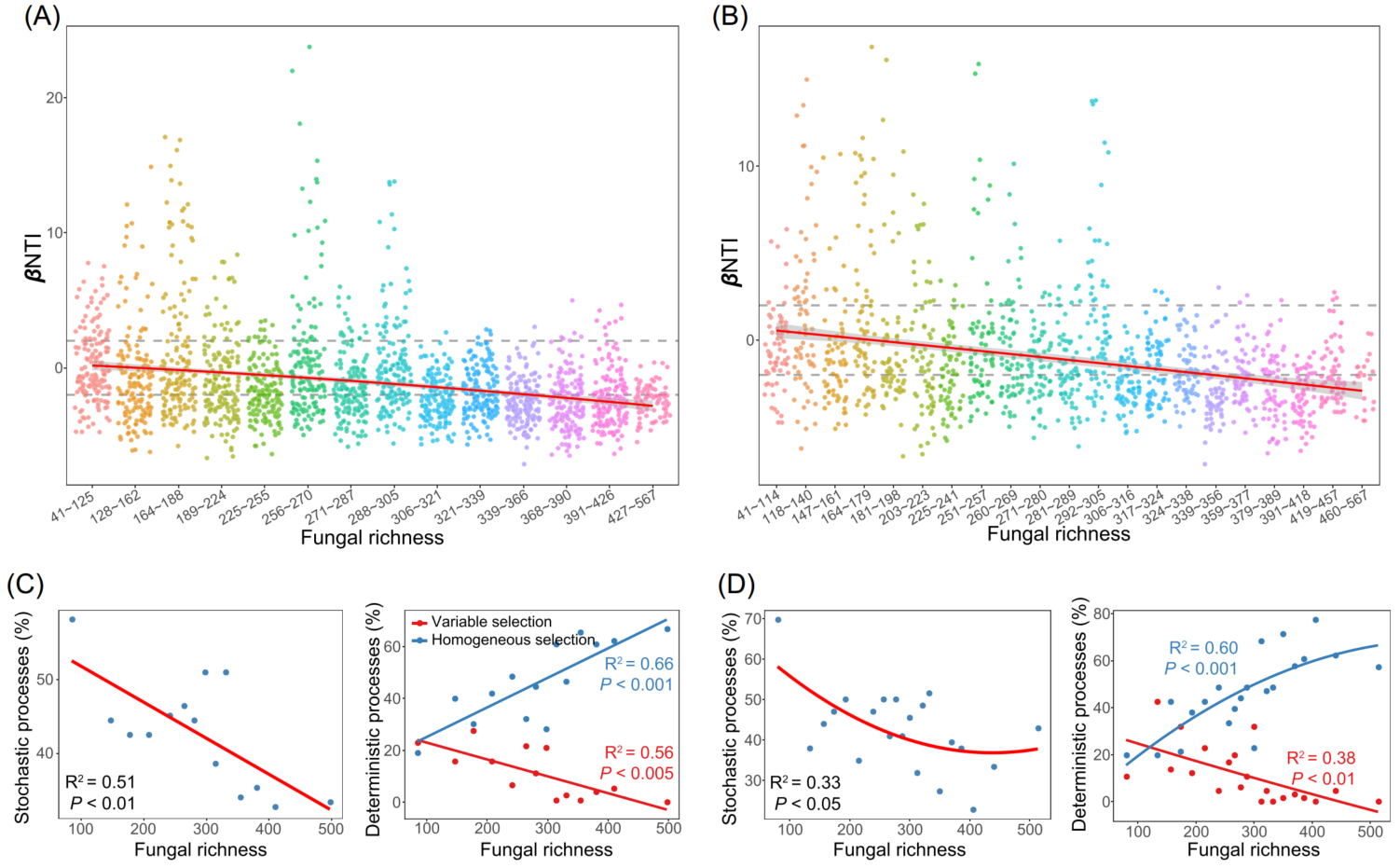
(A、B) βNTI在不同真菌丰富度分类中的分布模式,划分为14 (A)和21 (B)组;
(C、D) 水平虚线表示βNTI显著性阈值为+2和2。基于分成 14 (C)和21 (D)组的构建过程和真菌丰富度之间的关系,通过二阶多项式拟合的线性最小二乘回归分析估计。
为了进一步验证真菌丰富度对细菌群落组成的重要性,我们用独立的土壤样本进行了一个微宇宙控制实验。在中国西北地区的草地上采集土壤,利用抗真菌剂建立土壤真菌丰富度梯度,研究土壤真菌丰富度对细菌群落构建过程的影响以及特定细菌类群的变化。结果发现真菌的丰富度随着抗菌剂浓度的增加显著降低(图6A),表明真菌丰富度梯度形成的实验建立。在此背景下,细菌群落受随机过程的相对影响随着真菌丰富度的降低而增大,同质选择和变量选择的相对影响分别减小和增大(图6B),这与以上基于大规模调查的观察结果一致(图5)。细菌β-多样性在不同的真菌丰富度梯度间存在显著差异(图6C),且在不同的真菌丰富度梯度上存在显著的特定细菌类群(图6D)。而在真菌丰富度梯度中,细菌α-多样性没有显著差异。这一微宇宙研究结果为真菌丰富度在调节土壤细菌群落构建平衡过程中的重要作用提供了独立的实验验证。
图6. 将土壤真菌丰富度与细菌群落构建联系起来的的微宇宙研究
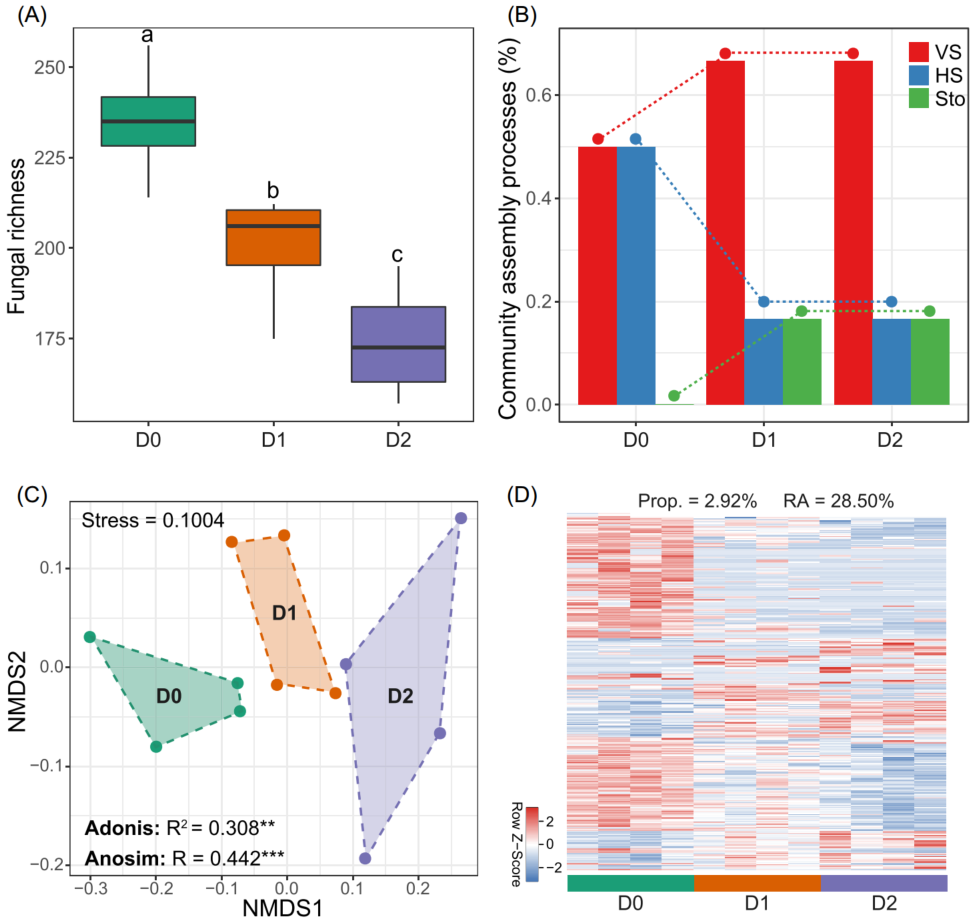
(A) 不同杀菌剂浓度(D0, no addition; D1, 6 mg kg-1; D2, 14 mg kg-1)处理形成土壤真菌丰富度的差异。不同的字母代表不同处理间的显著差异(P < 0.05; 与Kruskal-Wallis检验的多重比较);
(B) 通过确定性[同质选择(HS)和变量选择(VS)]和随机性过程(Sto)估计细菌群落在不同处理中的构建过程的变化;
(C) 显示不同处理间细菌β-多样性的非度量多维尺度(NMDS)排序图。使用Adonis和Anosim检验显著性( P < 0.01; * P < 0.001);
(D) 细菌类群的热图显示不同处理间相对丰度有显著差异。热图中的每一行都已标准化(均值为0,标准差为1),其颜色深度与分类群的标准化相对丰度成正比。Prop., 差异类群的比例;RA, 差异类群的总丰度。
讨论
揭示地下微生物群落的构建机制是生态学领域的一个重要课题,已在微生物生态学领域得到了广泛的探索。但很少有研究将生物作用与微生物β多样性的变化联系起来。在此基础上,探究了中国西北绿洲-荒漠过渡带不同生境和不同区域土壤细菌群落的潜在构建过程。通过大规模调查和微宇宙实验,我们为生物因子在塑造分布和调节细菌群落构建中起重要作用提供了有力证据。土壤真菌丰富度调节了土壤细菌群落构建过程的平衡,随着真菌丰富度的增加,随机过程逐渐减少。
由于气候变化,世界范围的干旱正在增加,并可能对旱地生态系统的结构和功能产生重大影响。在本研究中,我们观察到,在所有非生物(如土壤和气候)因素中,干旱对不同生境中细菌α-多样性变化的解释度最大,并且干旱的增加与细菌α-多样性的减少呈线性相关。这一结果得到了之前一项研究的支持,该研究报告了全球旱地的类似趋势。荒漠土壤细菌α-多样性受干旱影响最大,表明在全球干旱加剧的情况下,荒漠化会加剧土壤微生物多样性的损失。荒漠化是一个严重的问题,导致土地退化、生物多样性丧失和旱地生态系统功能减退。此外,AI对土壤微生物α-多样性的负影响在所有生境中均表现为下层比表层更强,说明干旱增加(如气候变化预测的干旱增加)时土壤深层微生物多样性的丧失更为显著。考虑到底层土壤包含全球50%的土壤有机碳储量和近35%的微生物生物量,深层土壤生物多样性的损失可能会极大地影响陆地生态系统的碳循环。以往的研究表明,干旱导致了多个生态系统属性的系统性和突变性变化,到2100年,超过20%的陆地表面将跨越干旱阈值。特别的是,我们的研究结果表明,为了减少干旱对陆地生态系统地下生物多样性和功能的负面影响,制定的政策不应忽视土地利用变化和敏感的下层土壤。
研究还发现,在低干旱环境中,微生物类群的共现频率更高(度更高),这也印证了降水越多,微生物相互作用越强的观点。在较低干旱水平下,网络复杂度较高,可能部分原因是降水增加刺激了生物量的增加,为不同物种之间的相互作用提供了更多的机会。此外,负关联与干旱之间存在显著的正相关,表明干旱的增加可能会引起更多拮抗或竞争的生物相互作用。这可能是由于高干旱环境的低湿度导致群落稳定性降低,物种间竞争加剧,与那些定殖在更孤立空间的物种相比降低了资源转移效率。在此基础上,我们发现干旱是影响细菌群落结构和物种共存的重要非生物因素。
物种间的相互作用决定了微生物群落的功能属性或生态位占有,在促进生态系统过程中发挥着重要作用。已有研究表明,物种间联系在驱动水稻土固氮营养菌群β多样性中起重要作用。与环境因素相比,生物因素更能预测土壤细菌的a-和β-多样性,表明生物相互作用对不同生境土壤细菌群落的多样性和构建具有更重要的作用。通过研究生物因素在决定群落中特定类群分布中的主要作用,也证实了这一观察结果。生物机制可能与物种关联有关,在生态学和生物地理学中,物种关联通常被用作群落中物种相互作用的代表。
由于适合度差异导致的物种间相互作用,如竞争和互惠等,会导致群落成员在环境异质性下的生态位划分。例如,有限营养源引起的竞争和物种间的拮抗作用限制了物种的共存,从而影响了微生物群落的构建。类群间的代谢相互依赖可诱导物种共存,导致微生物聚集。因此,这些研究可以支持我们的结论,即微生物类群之间的相互作用在大尺度上促进了不同生境群落β-多样性的显著变化,与我们的第一个假设相一致。
揭示地下微生物群落的构建机制对于更好地理解陆地微生物多样性的维持和产生具有重要意义。在本研究中,细菌群落βNTI值的两两比较与真菌丰富度差异显著相关,表明真菌丰富度与土壤细菌群落的随机和确定性构建过程的平衡密切相关。微宇宙实验进一步验证了土壤真菌丰富度与陆地生态系统细菌群落构建机制之间的联系。这与我们的第二个假设相对应。特别是随着真菌丰富度的增加,细菌群落受随机过程的影响相对减小,而受均匀选择和变量选择的相对影响分别增大和减小。这可能是由于土壤中真菌和细菌之间复杂的相互作用。例如,一些土壤真菌可以合成抗生素,并对细菌产生拮抗作用,或者土壤真菌可以分解难降解的有机物,如纤维素和木质素,为细菌共生利用提供基质。在这些情况下,真菌丰富度越高,细菌类群受同质选择的效应越强,其随机过程越弱,同质选择越强。我们的研究首先建立了土壤真菌丰富度与细菌群落构建机制之间的联系,并强调了跨界生物相互作用在调节复杂陆地生态系统微生物群落组装过程平衡中的潜在作用。
在本研究的范围内,应该考虑一些潜在的限制。首先,从跨界网络的拓扑性质推导出一些生物因素。相关性网络分析只是一个复杂系统的简单表示。此外,基于相关性的生态网络可能会产生虚假的结果,而且这些网络中类群之间的关联不能自动解释为相互作用。然而,生态网络信息仍然是观察群落成员拓扑属性的重要手段,并被认为是识别群落内物种关联的重要工具。其次,抗菌剂环已亚胺可能对所有真核生物都有毒性,环己胺可能也只作用于真菌的一个子集,可能选择与细菌相互作用更有利或显著的真菌成员。杀菌剂也可能作为细菌的资源影响其群落结构,微宇宙实验的狭小空间可能会限制细菌的迁移;这些因素都可能会影响细菌群落的构建。在未来的工作中,将在自然条件下进行更为系统的实验。
结论
我们的研究结果提供了有力的观测和实验证据,揭示了生物因素和干旱在塑造复杂陆地生态系统细菌地理分布和调节群落构建中的重要作用。土壤真菌丰富度调节土壤细菌群落构建过程的平衡,随着真菌丰富度的增加,随机组装过程逐渐减少;此结果在跨生物群落研究和控制实验中都得到证实的。气候变化导致干旱的增加会降低荒漠土壤和下层细菌的α-多样性,并引起更多拮抗或竞争的生物相互作用。我们的研究为将土壤真菌与干旱陆地生态系统的生物地理格局和土壤细菌群落构建机制联系起来迈出了重要的一步。考虑到跨界生物相互作用对群落构建的重要性,未来的实证和理论研究需要进一步探讨微生物多样性的产生和维持机制,以应对未来的气候干旱变化。这与其他不利影响(例如,水资源供应减少)一样,可能对世界各旱地的粮食生产等关键生态过程和服务构成严重威胁。
方法
土壤样品采集
共选取农田37处,森林28处,湿地15处,草地26处,荒漠20处共126处。采样地点在中国的西北沿河西走廊,从36°56’N到40°34’N,从94°37’E 到103°31’E (横断面间隔1257.6公里) (图1)。这些栖息地的优势种包括Zea mays (农田)、 Calligonum spp., Stipa spp., Leymus spp., and Achnatherum spp. (湿地、草原和沙漠) 和Populus spp.、 (森林)。土壤类型为亚纹石和钙石,结构疏松,有机质含量低。
在2017年7月至8月(地上植物生物量的最高时期),在每个站点采集了3个100 m2的样地。每个地块合并5个土壤芯(直径为2.5 cm),分别在0-15 cm和15 - 30 cm深度采集。一个沙漠地下样本因DNA提取失败而被舍弃。因此,本研究共有251个土壤样品。
土壤理化的测定:采用标准方法测定土壤pH、水分、阳离子交换能力(CEC)、有机碳(SOC)、溶解有机碳(DOC)、全氮(TN)、硝态氮(NO3)、铵态氮(NH4)、全磷(TP)、速效磷(AP)、全钾(TK)、速效钾(AK)。从Worldclim数据库(www.worldclim.org)获得了所有采样点的气候数据,包括年平均温度(MAT)。此外,利用全球潜在蒸发数据库估算了每个站点的干旱度(AI, 1 -降水/蒸腾),该数据库是基于WorldClim提供的。
使用土壤FastDNA SPIN试剂盒(MP Biochemicals, Solon, OH, USA)从土壤样品中提取总基因组DNA。采用高通量扩增子测序技术测定分别对土壤细菌16S rRNA基因的V4-V5区(5F/907R)真菌18S rRNA基因的ITS1区(ITS 5-1737F/ITS2-2043R)进扩增。测序在Illumina HiSeq2500平台上进行(Illumina Inc., San Diego, USA)。使用DADA2 v1.14将经过质量过滤的reads组装成扩增子序列变体(ASVs)。当ASV出现在少于两个样本时,就过滤掉。分类学使用核糖体数据库项目分类器(Ribosomal Database Project Classifier)工具进行序列鉴定,使用SILVA数据库(138版)和UNITE+INSD (UNITE和国际核苷酸序列数据库)数据库分别对细菌和真菌进行比对。在计算土壤微生物多样性之前,对微生物群落数据进行抽平,以达到最小的序列数,细菌为28,955,真菌为20,065。计算细菌的香农多样性和真菌的丰富度,这是最广泛使用的。真菌群落以子囊菌门(相对丰度68.8%)为主,担子菌门(相对丰度10.8%)、被孢菌门(相对丰度9.2%)和壶菌门(相对丰度5.8%)次之。野外调查研究的原始测序数据保存在大数据中心基因组序列档案库,生物项目编号为PRJCA004036。
微宇宙控制实验
在独立于上述大规模调查的土壤中进行微宇宙实验,解释了这两项研究之间的微小方法差异,并使我们能够测试土壤真菌丰富度和细菌群落构建之间的关系,用于评估空间格局的数据。2019年8月,在中国西北干旱区(38°18’N, 100°9’E)进行土壤采样,采样深度为0-15 cm。该地点为沙棘属(Calligonum spp)为主的草地,其干旱条件与野外调查地点相似(干旱度=0.663)。
对土壤样品进行了<2毫米的过筛,移除石头和碎片,并放置在人工气候室(25摄氏度)几个星期,以达到平衡和稳定的状态。将制备好的土壤样品移入塑料盆中,按照前人研究方法,用杀菌剂(环己亚胺)溶液进行修正,使最终浓度为0 (D0)、6 mg kg-1 (D1)和14 mg kg-1 (D2)。杀菌剂的浓度是根据标准生态毒理学规范确定的,以确定农药可能的环境效应。共准备12个微宇宙(每个500克;3个处理,4个重复)。微宇宙中的水分含量被调整到60%的保水能力,以便在孵育期间保持微生物活动(如有需要可添加无菌水)。微宇宙上的几个孔(1毫米)被铝箔覆盖,以避免污染,并转移到人工气候室在20℃的黑暗环境中培养60 d。孵育后,采集土壤样品进行多样性测定,并按照上述方法对土壤细菌和真菌群落进行分析,进行跨生物群落研究。在计算土壤微生物多样性之前,对微生物群落数据进行抽平,以达到最小的序列数,细菌为55,975,真菌为26,157。计算已抽平ASVs表中细菌的Shannon多样性和真菌的丰富度。微宇宙研究的原始测序数据保存在BIG数据中心基因组序列档案库,生物项目编号为PRJCA004037。
数据分析
为了预测采样区域内细菌Shannon多样性的分布,采用了考虑环境变量影响的co-kriging插值方法。kriging模型包含了5个环境预测因子:土壤性质(土壤C和pH)、气候(AI和MAT),因为这些变量的高分辨率信息在全球尺度上是可用的。该网格的土壤性质信息通过ISRIC(全球网格化土壤信息)土壤网格获得(https://soilgrids.org/#!/?layer=geonode:taxnwrb_250m)。这一分析在R中的automap包进行,该包通过自动估计半变异函数并执行kriging来自动完成插值过程。地图的交叉验证是基于每个采样点的预测值和观测值之间的皮尔逊相关性,使用automap包中的autoKrige.cv。
为了评估物种在不同生境和区域的共存情况,构建了细菌和真菌跨界共现网络。为了降低数据库稀有ASVs,我们集中研究了存在于所有土壤样本中超过10%的微生物类群。与斯皮尔曼相关系数(ρ) >0.6或<-0.6同时P < 0.001被用于构建网络,该网络在文献中被广泛使用,在研究中具有可比性。我们计算了检测到土壤和气候因子的各站点的平均值,然后根据每个站点该分类单元的相对丰度对其进行加权。这些被认为是每个类群偏好的环境条件,类似于生态位空间。估算了环境偏好性的值和节点度之间的Pearson相关性。此外,使用igraph包中的induced_subgraph函数,提取每个土壤样本的子网络。计算每个样本子网络的拓扑参数,包括平均度(AD)、负关联比例(Neg)和细菌与真菌类群相互关联比例(Int),以估计潜在的生物相互作用,这种相关作用在研究细菌α-和β-多样性变化的贡献时被视为生物因子。平均度是指群落中物种的连通性。细菌和真菌类群之间的负相互作用比例和互作比例可以反映其潜在的生物学相互作用。网络可视化是使用交互式Gephi平台(https://gephi.org)。
计算DDRs作为地理距离与群落相似性(1 - Bray-Curtis度量的不相似度)之间关系的最小二乘回归的斜率。使用R中的vegan包,采用标准和偏曼特尔检验评估环境、生物和地理变量对细菌群落结构的影响。
利用Stegen等人描述的框架进行零模型分析,将群落构建潜在驱动机制划分为确定性过程和随机性过程。利用基于零模型的系统发育β-多样性指标(βNTI)分别测定系统发育和类群多样性的差异。使用phangorn软件包,通过bootstrap分析(100个重复)推断出一个邻接(NJ)系统发育树。βNTI <-2表示系统发育周转显著低于期望值(即同质选择);相反,βNTI>2表示系统发育周转显著高于期望值(即变量选择)。|βNTI|<2表示随机过程主导。利用曼特尔检验探究影响土壤细菌群落组装过程的主要因素。将βNTI值与每个变量的欧几里德距离矩阵关联起来,评估了源于变量梯度的群落构建过程中的变异。采用999次置换来确定这些差异的统计学显著性,以及R中的vegan包mantel函数进行分析。
采用随机森林(R中rfPermute包的rfPermute函数)和多元线性回归模型(R中的stats包的lm函数)与方差分解(R中relaimpo包的calc.relimp函数)来评估对于拓扑参数影响因素的重要性。采用基于距离的线性模型和Bray-Curtis距离矩阵的前向选择方法,通过评估解释变量(R2)检验环境变量对β-多样性的显著性和重要性。这些结果由主坐标分析(CAP)得到。此分析是使用vegan包的ordiR2step和capscale函数进行的。利用R中ecodist包的曼特尔函数,采用标准和偏曼特尔检验来评估环境、生物和地理变量对细菌群落结构的影响。利用vegan包的metaMDS函数,采用非度量多维尺度(NMDS)分析来可视化不同组间的样本关系。
-
点赞 (0人)
- 收藏 (0人)

