
iMeta|同济大学朱瑞新团队靶向基石菌种可恢复非酒精性脂肪肝中失调的产丁酸菌
- 看不见的线
- 3435
- 2024-06-04 14:57:20
- 文章来源:iMeta
靶向基石菌种可恢复非酒精性脂肪肝中失调的产丁酸菌

https://doi.org/10.1002/imt2.61
RESEARCH ARTICLE
●2022年11月16日,同济大学朱瑞新团队等在iMeta在线发表了题为“Targeting keystone species helps restore the dysbiosis of butyrate-producing bacteria in non-alcoholic fatty liver disease”的文章。
●产丁酸菌的生态失调是非酒精性脂肪性肝病发展的关键因素之一,该研究发现多个基石菌种可有效地使NAFLD的微生物组成向正常肠道菌群恢复,提示了一种新的潜在的NAFLD微生物治疗方法。
● 第一作者:吴顶峰;刘蕾;焦娜
● 通讯作者:朱瑞新(rxzhu@tongji.edu.cn);Rohit Loomba (roloomba@ucsd.edu);Lixin Zhu (朱立新)(zhulx6@mail.sysu.edu.cn)
● 合作作者:张一达,杨莉,田川,兰平
● 主要单位:浙江大学医学院儿童医院,国家儿童临床医学研究中心;同济大学生命科学技术学院,上海第十人民医院;中山大学附属第六医院,广东省胃肠病研究所;美国加州大学圣地亚哥分校,医学系胃肠病学和流行病学NAFLD研究
亮 点
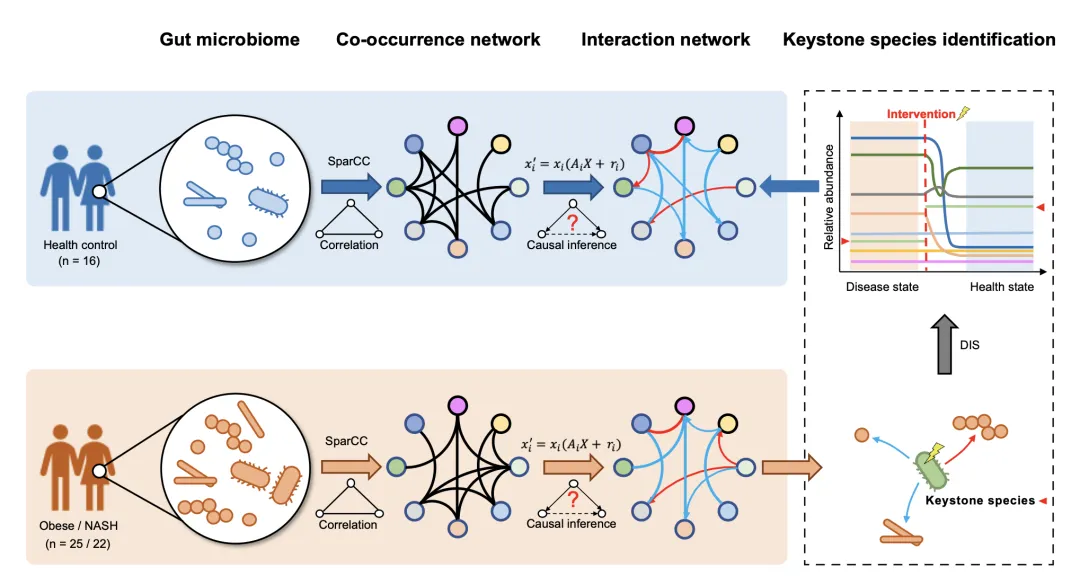
●产丁酸菌的生态失调是NAFLD发展的关键因素
●将生态学理论与动态干预模拟相结合的因果算法可以从宏基因组数据中挖掘微生物基石菌种
●以Porphyromonas loveana、 Alistipes indistinctus和Dialister pneumosintes为代表的NASH的基石菌种,为NAFLD治疗提供了潜在的精准干预策略
摘 要
肠道菌群失调是非酒精性脂肪性肝病(NAFLD)的致病因素之一,也影响其治疗和干预。在肠道菌群中,调节生态群落的完整性和稳定性的基石菌种已成为NAFLD的潜在干预靶点。本研究收集了来自纽约的22名非酒精性脂肪性肝炎(NASH)患者、25名肥胖患者和16名健康个体的粪便样本,进行16S rRNA基因测序,并基于因果推理理论和动态干预模拟(DIS)建立了一种疾病基石菌种识别算法。算法与结果在来自加州的独立队列中进行外部验证。本研究在NAFLD肠道中鉴定出以Porphyromonas loveana、Alistipes indistinctus和Dialister pneumosintes为代表的8个基石菌种,可有效地使NAFLD的微生物组成向正常肠道菌群恢复,恢复率为92.3%。功能分析表明这些基石菌种通过调节肠道氨基酸代谢和酸碱环境,促进了NAFLD患者中显著减少的产丁酸菌的生长,主要包括Lachnospiraceae和Ruminococcaceae的菌种。我们的研究结果表明了基石菌种在恢复正常肠道微生物微生物组成方面的重要性,提示了一种新的潜在的NAFLD微生物治疗方法。
视频解读
Bilibili:https://www.bilibili.com/video/BV18t4y1N7N6/
Youtube:https://youtu.be/E0PMO5MexiQ
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
非酒精性脂肪肝(NAFLD)是一种复杂的多因素疾病,其发病机制尚不明晰。最近对代谢性脂肪肝的定义强调NAFLD是从肥胖到代谢综合征和糖尿病的连续变化过程,其中非酒精性脂肪性肝炎(NASH)是伴有肝脏炎症的NAFLD的晚期形式。研究表明,肠道菌群的失调影响NAFLD的发生和发展。前期,利用肠道微生物组的全基因组测序数据,我们已经鉴定出了37种可区分轻、重度NAFLD患者的微生物标记物。此外,我们还观察到,微生物产生的代谢物,包括内源性乙醇、胆汁酸和氨基酸,参与了NAFLD的发病机制。许多针对肠道菌群的微生物干预疗法,如益生元、益生菌和粪菌移植(FMT)等正在被考虑用来治疗NAFLD 。然而,这些微生物干预手段并没有达到令人满意的效果,这可能由于我们对肠道中生态系统的结构复杂性的片面解读。因此,进一步研究疾病发展过程中微生物群的动态变化及其变化背后的机制,对于NAFLD精准治疗和确定其他相关疾病的微生物靶点至关重要。
基石菌种是维持和稳固微生态系统所需的重要菌种,是基于肠道微生物组的干预的优秀候选靶点。基石菌种已经在一些疾病中报道,如牙周病和艰难梭菌感染。基石菌种的改变可以通过生态系统成员之间的相互作用来引起整个群落的动态变化。因此,我们假设,与FMT不同,针对基石菌种的治疗可以准确地控制肠道菌群的状态,是NAFLD的一个有效的精准干预策略。
二代测序技术的发展使我们能够在真实环境中研究微生物组。相关分析已成为研究微生态系统相互作用的常用方法,基于共现网络的拓扑结构,定义网络的枢纽节点(Hub),并将其视为基石菌种。然而,受混杂因素的影响,共现网络并不能准确地反映微生物之间的因果关系,限制了疾病基石菌种鉴定的准确性。这些限制可以通过因果推理分析来解决。我们注意到因果理论在各个研究领域得到了重视和发展,并已经被应用于微生物相关的研究中。与相关分析相比,因果理论更有利于构建微生态系统的动态模型,从而确定基石菌种。
在这里,我们收集了来自纽约的22名NASH患者和16名健康个体的粪便样本,用于16S rRNA基因测序。采用与生态学理论相结合的因果算法构建微生态系统的动态模型,并通过动态干预模拟(DIS)确定基石菌种(图1A)。基于这一策略,我们能够表征肠道微生态系统的组成和相互作用的变化,从而描述肠道菌群在NAFLD中的潜在作用,并确定疾病精准干预的潜在靶点。该策略进一步在25名肥胖患者和来自加州的外部验证队列中进行应用和分析比较。
结 果
肥胖和NASH患者中肠道微生物的丰度变化
本研究对16名健康对照组、25名肥胖患者和22名非酒精性脂肪性肝炎(NASH)患者的粪便样本进行了16S rRNA基因测序,获得了肠道微生物组的定量丰度数据。基于因果理论,建立了对照组和疾病组微生态系统的动力学模型。然后通过动态干预模拟(DIS)确定NASH的基石菌种,以指导NASH的治疗(图1A)。
与正常对照组相比,差异分析在NASH患者的肠道中发现了39个差异菌种(图1B)。这些差异菌种主要属于Clostridiales,包括Family XI、Lachnospiraceae、Ruminococcaceae、Peptostreptococcaceae和Family III等。在NASH中,Family XI的差异菌种丰度显著上调,而Lachnospiraceae和Ruminococcaceae的差异菌种丰度下调(图1B)。在肥胖症中也观察到了类似的变化。有趣的是,在从正常发展到肥胖再到NASH的过程中,肠道菌群中差异菌种的丰度发生渐进式的变化。此外,这些差异菌种对正常和NASH样本具有较强的分辨能力,例如Porphyromonas loveana、Alistipes indistinctus和Dialister pneumosintes的AUC分别为0.794、0.766和0.832。
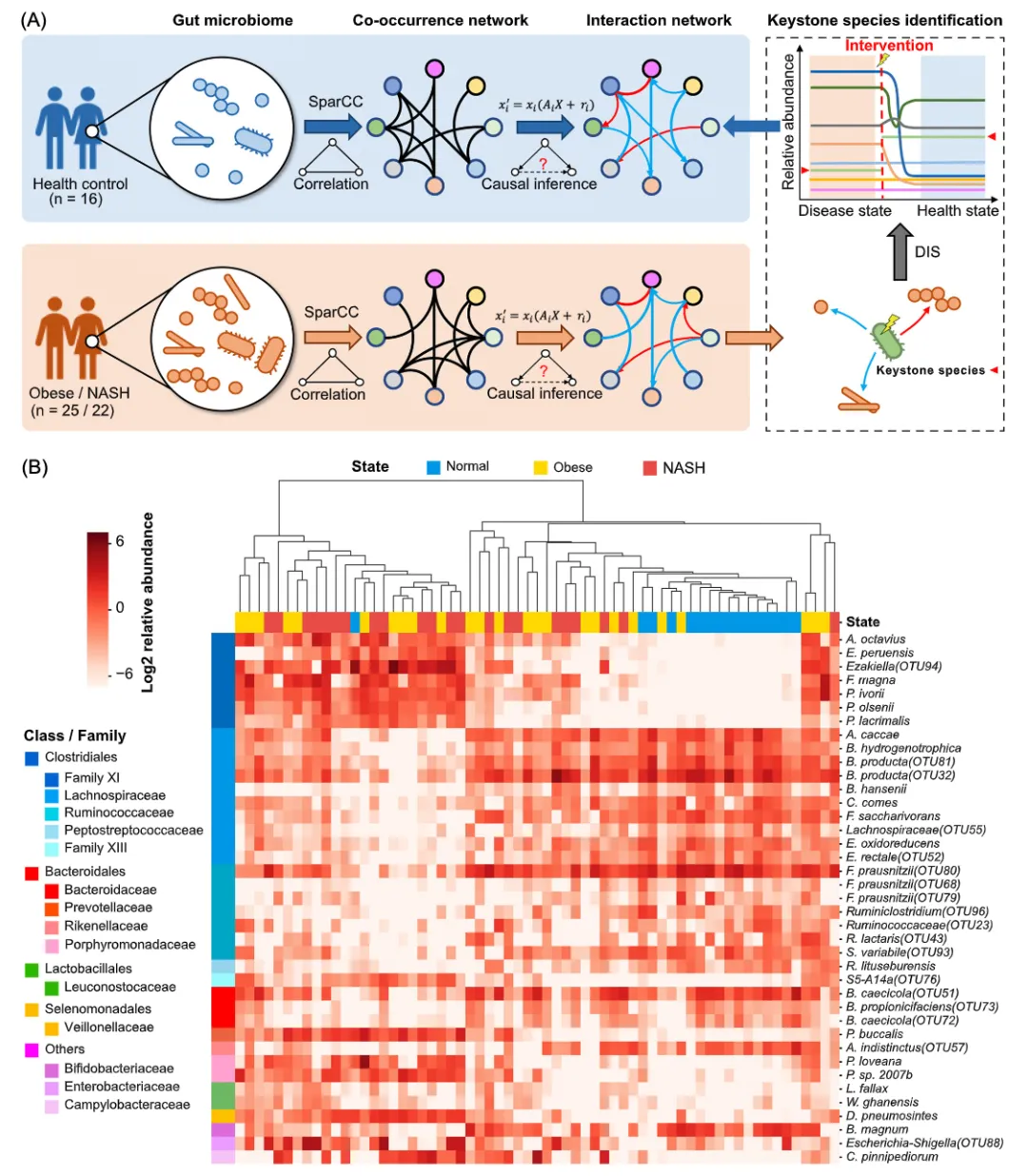
图 1.本研究分析流程和差异微生物的丰度变化
(A)基于丰度数据和微生物共现网络,利用与生态学理论相结合的因果推理算法,构建了微生态系统的动力学模型。然后,通过动态干预模拟(DIS),确定了有助于恢复肠道微生态系统生态失调的基石菌种;(B)39个菌种在正常对照组和非酒精性脂肪性肝炎(NASH)疾病组之间表现出不同的丰度,这些差异菌种大多数属于Clostridiales。在NASH中,Lachnospiraceae和Ruminococcaceae的差异菌种表达下调。在肥胖症中也观察到了类似的变化。
正常、肥胖和NASH受试者肠道中不同的微生物相互作用模式
利用因果推理算法,本研究分别构建了正常、肥胖和NASH组的肠道微生物互作网络(图2A-C),并基于网络拓扑属性确定每组的枢纽菌种(Hub species,图2D-F)。
正常、肥胖和NASH组的肠道菌群表现出不同的微生物互作模式(图2)。在正常组中,微生物互作网络表现出一种异质性模式(幂律分布 R2 = 0.70),这是一个典型的无标度网络的特征(图2A)。Lachnospiraceae(Positive/Negative= 58/47)、Ruminococcaceae(P/N = 20/10)和Bacteroidaceae(P/N = 52/33)中的枢纽菌种主要对网络产生正调控影响,而那些属于Family XI(P/N = 0/38)的枢纽菌种主要对正常肠道微生物产生负调控影响(图2G)。
与正常组相比,肥胖和NASH肠道内的微生物相互作用表现出较大的差异。网络的菌种互作分布相对均匀,菌种之间互作关系更多(图2B、C)。虽然Lachnospiraceae、Family XI、Bacteroidaceae仍是网络的枢纽菌种,但Prevotellaceae、Veillonellaceae、Peptoniphilus、Porphyromonadaceae等科中的菌种被鉴定为肥胖和NASH的枢纽菌种。肥胖和NASH特定枢纽菌种包括Prevotella buccalis、A. indistinctus、Porphyromonas sp. 2007b和D. pneumosintes(图2D-F)。
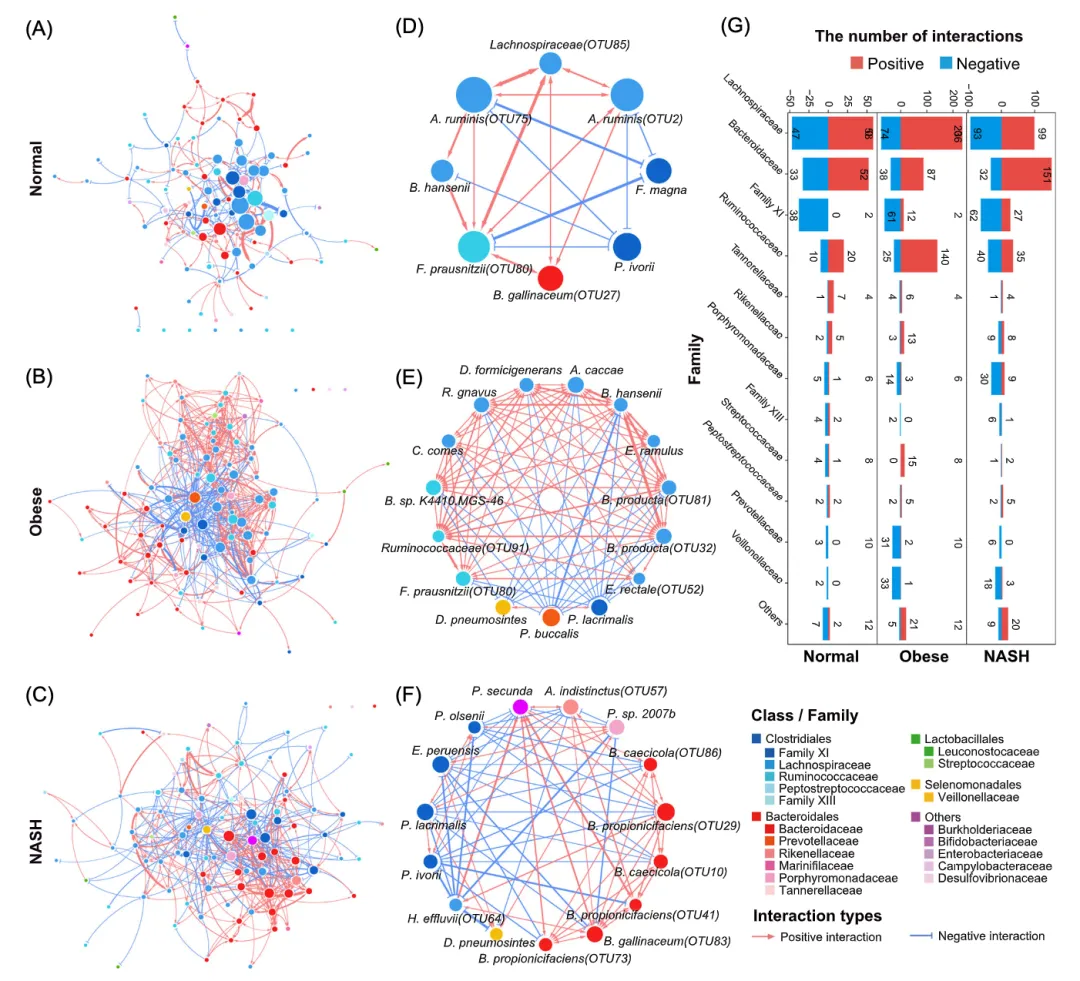
图 2.正常、肥胖和非酒精性脂肪性肝炎(NASH)受试者肠道中的微生物互作网络和枢纽菌种
正常(A)、肥胖(B)和NASH (C)组的微生物互作网络,以及正常(D)、肥胖(E)和NASH (F)组的枢纽菌种的相应子网络。节点的颜色表示菌种的类别,边的颜色表示每两个菌种之间的正(红色)或负(蓝色)的相互作用。(G)正常、肥胖和NASH组的微生物互作网络中各科的相互作用分布
NASH的基石菌种驱动疾病肠道菌群向正常菌群的变化
基石菌种的丰度的改变可能会引起其他菌种的变化,并深刻地影响肠道微生态稳态。因此,本研究采用DIS来模拟微生物干预后肠道菌群的动态变化,并采用迭代特征消除(IFE)来识别那些对整个微生物群总体影响最大的菌种。
根据DIS分析(图3A),由于处于网络拓扑中心位置,枢纽菌种在肠道微生态系统中具有较高的影响力。针对肥胖患者的枢纽菌种的干预,如P. buccalis(干预评分IS = 0.397)、Ruminococcus torques(IS = 0.288)、Blautia hansenii(IS = 0.284)、Anaerostipes caccae(IS = 0.281)和D. pneumosintes(IS = 0.279)会导致肠道菌群结构发生显著变化,从而恢复正常的肠道菌群结构。类似地,对于NASH肠道菌群,针对枢纽菌种如P. sp. 2007b(IS = 0.376)、D. pneumosintes(IS =0.294)、Peptoniphilus lacrimalis(IS =0.289)、Agathobacter ruminis(IS =0.254)和Ezakiella peruensis(IS =0.247)的干预能够使肠道菌群结构发生显著变化,从而恢复正常的肠道菌群结构。相对于枢纽菌种,其他菌种对肠道菌群的影响较小。
针对多个菌种的干预比针对单一菌种能够产生更好的效果。因此,我们整合DIS和IFE算法开发一个新的算法用于确定具有最高干预潜力的基石菌种组合。我们分别从肥胖和NASH肠道微生物中鉴定出11个和8个基石菌种,组合干预分数(CIS)分别为0.903和0.923(图3)。这表明针对这些基石菌种的干预,患者肠道菌群可以最大限度地恢复到正常的菌群结构(图3B)。针对NASH中的8个基石菌种的干预会导致正常菌群的枢纽菌种Lachnospiraceae和Ruminococcaceae的丰度增加(图3B)。同时,NASH菌群的枢纽菌种也对干预有反应,并且大部分差异菌种的丰度向正常状态变化。
在8个NASH基石菌种中,前3个基石菌种,P. loveana、A. indistinctus 和D. pneumosintes表现出较强的干预能力(CIS = 0.664),其他5个基石菌种起次要作用(图3A)。同样,肥胖的前3个基石菌种也表现出较强的干预能力(CIS = 0.587)。NASH中,前3个基石菌种为正常或疾病微生物互作网络的枢纽菌种(P < 0.05),在NASH患者进行微生物干预时引起重大变化(图3C)。NASH中P. loveana的丰度升高,因此我们在干预时降低该菌种的丰度。降低P. loveana的丰度主要增加了Lachnospiraceae菌种的丰度,其中大多数为正常菌群的枢纽菌种。NASH中 A. indistinctus的的丰度显著减少。因此,通过干预操作升高A. indistinctus的丰度,并导致Bacteroidaceae菌种丰度升高和Family XI菌种丰度减少。NASH患者的D. pneumosintes含量显著增加,因此干预操作降低D. pneumosintes的丰度。该干预将导致Lachnospiraceae和Ruminococcaceae菌种的丰度增加。对这三个基石菌种的同时干预可以恢复许多肠道微生物的丰度,特别是Lachnospiraceae和Ruminococcaceae(两者在正常菌群中都是丰富存在的科),从而促进正常肠道菌群的重建(图3C)。
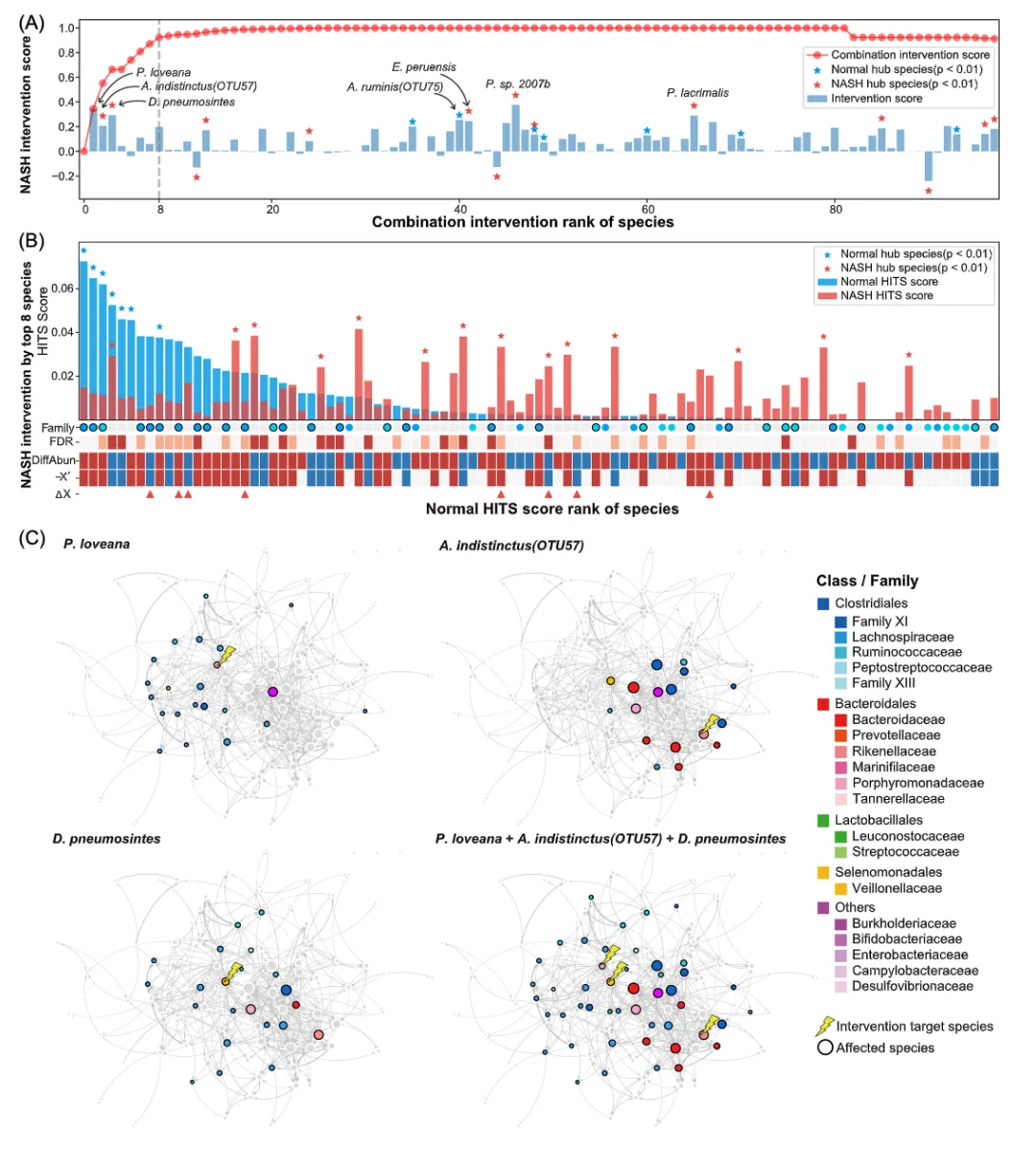
图 3.非酒精性脂肪性肝炎(NASH)微生物群的动态干预模拟(DIS)
(A)对肠道微生物的干预评分(IS)。每个微生物的IS显示在条形图中,蓝色和红色的星星分别表示超链接引导主题搜索(HITS)在正常对照组和NASH枢纽菌种中的重要性得分。IS负值表明菌群结构向与健康对照组差距更大的状态变化。红色曲线表示DIS依次选择的微生物的组合干预得分(CIS),表示多个菌种同时干预后的干预效果。前8个达到CIS大于0.9的基石菌种,用虚线标注出。(B)微生物干预对NASH微生物群的影响,其中前8个基石菌种来自(A)。条形图为正常微生物群中菌种的HITS得分排序,枢纽菌种用星形标记。在Family轴上标记了Lachnospiraceae(蓝色)和Ruminococcaceae(浅蓝色),黑色边框表示干预后丰度恢复到正常水平。DiffAbun:丰度从正常状态变化为NASH(红色:增加;蓝色:减少),错误发现率(FDR)(红色:FDR < 0.01;浅红色:FDR < 0.05)。:干预后即时微生物丰度变化的负性表征(红色:> 0;蓝色:< 0)。在中,用三角形标记了8个需要干预的基石菌种。(C)前3个基石菌种(Porphyromonas loveana,、Alistipes indistinctus 和Dialister pneumosintes)的干预效果。节点的黑色边框表示有显著的干预效果。
基石菌种调控NASH肠道菌群的潜在作用机制
P. loveana、A. indistinctus和D. pneumosintes被确定为NASH微生物组中最重要的基石菌种,对NASH菌群的干预能力最高。为了进一步了解这些基石菌种导致菌群变化作用机制,我们使用PICRUST2获得了它们的基因信息,并进行了功能(KEGG Module)富集分析(图4A)。
如图4A所示,基石菌种P. loveana的基因组严重缺乏氨基酸生产基因,而辅助因子和维生素生产基因含量丰富。氨基酸,如谷氨酸、甘氨酸、丙氨酸、酪氨酸、天冬氨酸、缬氨酸等,是生产辅助因子和维生素所必需的底物,也可用于生产短链脂肪酸。因此,P. loveana丰度的增加会导致NASH的氨基酸消耗增加。在基石菌种D. pneumosintes中也观察到类似的基因富集模式,表明D. pneumosintes可能导致NASH中氨基酸消耗的增加。
基石菌种A. indistinctus参与了氨基酸的生产,包括丝氨酸、苏氨酸、缬氨酸、异亮氨酸、亮氨酸、精氨酸、脯氨酸、谷氨酸和组氨酸等(图4A)。A. indistinctus的丰度下调(FDR = 0.020)表明微生物合成的氨基酸减少。
更重要的是,D. pneumosintes编码F型ATP酶复合物的8个基因(图4A,B)。这表明D. pneumosintes可以利用肠腔内的H+合成ATP,这会引起了Lachnospiraceae和Ruminococcaceae菌种的丰度变化(图3C)。而相关分析表明D. pneumosintes与Lachnospiraceae和Ruminococcaceae丰度呈负相关(P < 0.01,图4C)。既往研究表明,Lachnospiraceae和Ruminococcaceae菌种对环境pH非常敏感,并影响其丁酸生产能力。因此,NASH中D. pneumosintes丰度的增加(FDR = 0.004)可能会肠道pH改变,从而降低Lachnospiraceae和Ruminococcaceae的丰度和丁酸的产量。
总的来说,虽然相对丰度较低,但P. loveana, A. indistinctus和D. pneumosintes能够调节肠道Lachnospiraceae和Ruminococcaceae菌种丰度,揭示了它们在NASH肠道微生态系统中的关键作用。
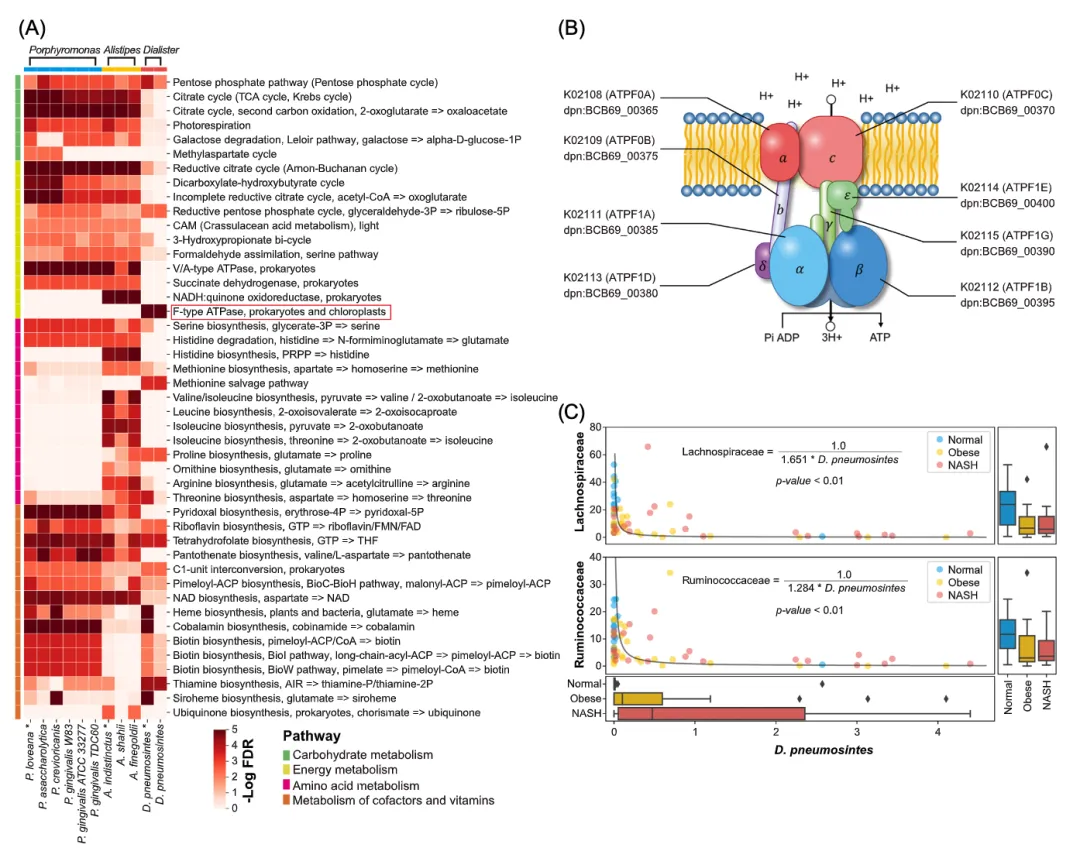
图 4.基石菌种的功能富集分析
(A)利用PICRUST2进行基因(KO)注释,对Porphyromonas loveana、Alistipes indistinctus和Dialister pneumosintes进行KEGG模块富集分析。模块根据KEGG通路进行分类,并用不同的颜色进行标记。热图绘制了富集的重要性分数(-log false discovery rate [FDR]);(B)D. pneumosintes 基因编码F型ATP酶复合物的全部8个基因。这种蛋白质复合物可以利用膜质子梯度来产生ATP;(C)D. pneumosintes的丰度变化与Lachnospiraceae 和Ruminococcaceae的丰度变化显著相关,均具有显著的函数关系。箱形图为Lachnospiraceae、Ruminococcaceae 和D. pneumosintes在正常、肥胖和非酒精性脂肪性肝炎疾病组肠道中的丰度分布。
基于独立队列验证NAFLD基石菌种的鉴定过程
我们在NAFLD验证队列中观察到类似的微生物变化模式。简而言之,Lachnospiraceae和Ruminococcaceae菌种在NAFLD患者肠道中的丰度显著降低(P < 0.01)。此外,发现队列中NASH微生物组的基石菌种A. indistinctus也显著下调。这些差异菌种也表现出较强的样本识别能力,AUC 为0.894。
同样,Lachnospiraceae和Ruminococcaceae是正常肠道菌群的枢纽菌种,在维持正常微生物群落的稳态中起着关键作用。
从验证队列中鉴定出的基石菌种与从发现队列中鉴定出的基石菌种部分重叠。这两个队列的共有的基石菌种包括Blautia producta、Bacteroides barnesiae和A. caccae。同样,针对这些基石菌种进行微生物干预,能够恢复Lachnospiraceae和Ruminococcaceae菌种的丰度。
讨 论
在本研究中,我们基于因果理论和动态干预模拟(DIS),开发了一种新算法应用于肠道菌群的基石菌种识别。我们鉴定了非酒精性脂肪性肝炎(NASH)以P. loveana、A. indistinctus和D. pneumosintes 为代表的基石菌种组合,表现出对NASH肠道微生物的干预潜力。
青少年(发现队列)和成人(验证队列)非酒精性脂肪肝(NAFLD)的肠道菌群最突出的特征是Lachnospiraceae和Ruminococcaceae菌种的丰度降低,它们是Clostridiales的两个优势科。作为肠道中主要的丁酸产生细菌,Lachnospiraceae和Ruminococcaceae能够通过丁酸刺激黏膜中的T调节细胞来抑制肠道炎症,从而抑制NASH 的发病。这两类细菌在结构和功能上的重要性使它们成为微生物干预NAFLD的理想靶点。
在NASH菌群中,基石菌种,特别是P. loveana、A. indistinctus和D. pneumosintes 可以迅速改变Lachnospiraceae和Ruminococcaceae菌种的丰度,使微生物组成恢复到正常的肠道菌群群状态。这些菌种能够通过发酵产物影响其他群落成员。NASH中P. loveana升高,将其从NASH菌群中移除进行干预。P. loveana丰度减少导致氨基酸消耗减少,为Lachnospiraceae和Ruminococcaceae等其他细菌的生长留下了更多的资源。A. indistinctus在NASH中丰度降低,因此在NASH菌群中增加其丰度进行干预。A. indistinctus具有许多与氨基酸合成相关的基因。除了支持肠道菌群的蛋白质合成外,肠道中产生的这些氨基酸还可以作为短链脂肪酸(SCFA)生产的微生物发酵底物,如由苏氨酸、赖氨酸和谷氨酸合成丁酸。
因此,增加A. indistinctus丰度不仅可以通过其氨基酸生产促进微生物群落其他成员的生长,还可以促进SCFA生产,维持肠道免疫稳定。NASH中D. pneumosintes 升高,将其从NASH微生物组中移除进行干预。D. pneumosintes 编码F型ATP酶复合物的8个基因,这些基因可以利用肠道环境中的质子合成ATP,有助于维持低肠道pH,从而促进产丁酸的Lachnospiraceae和Ruminococcaceae菌种的生长。
与基石菌种相比,NASH患者Lachnospiraceae和Ruminococcaceae菌种的丰度改变更加明显,但他们没有表现出更好的干预能力,即便通过迭代特征消除(IFE)也无法被认定为基石菌种。我们的研究结果表明,基石菌种可能不是微生物群落中最丰富的菌种,因此靶向疾病中主要的丰度改变菌种可能不是一种有效的干预策略。相比之下,P. loveana、A. indistinctus和D. pneumosintes在IFE中干预打分超过其他基石菌种。虽然它们并不是疾病中最主要的差异菌种,但它们在恢复正常肠道菌群方面表现出最高的潜力。它们对NASH的微生物干预能力可能归因于其代谢产物对微生物群落的其他成员的深远影响。这些广泛的影响使它们在维持正常微生物群落的完整性和稳定性方面起到特殊作用。
虽然重建复杂系统的互作网络非常困难,特别是对于横断面数据,但我们提出了一种基于因果推理的微生态系统建模方法,该方法得益于先验知识,提高菌种互作发现的准确性。然后根据DIS的定义确定了基石菌种,丰富的先验知识和严格的方法框架保证了本研究的可靠性以及后续实验和临床治疗的可行性。
结 论
总之,我们通过因果推理和动态干预模拟,从横断面微生物组数据中确定了微生物基石。我们从非酒精性脂肪性肝病(NAFLD)肠道中鉴定出以P. loveana、A. indistinctus和D. pneumosintes 为代表的基石菌种,可以有效地调节NAFLD的微生物组成向正常的肠道菌群改变,特别是Lachnospiraceae和Ruminococcaceae的菌种。通过一个独立的NAFLD队列验证,我们的研究结果提示了一种新的NAFLD微生物治疗策略。此外,本研究所描述的微生物基石菌种鉴定策略可能有利于医学、环境科学和微生物学等多个领域的微生物组学研究。
方 法
发现队列
在我们之前的研究中描述了来自纽约的发现队列。原始测序reads和相关的元数据可从MG-Rast(http://metagenomics.anl.gov/linkin.cgi?project=1195)下载。发现队列包括了22名经活检证实的NASH患者、25名肥胖患者和16名健康对照者的青少年粪便样本,并使用454-FLX-Titanium Genome Sequencer (Roche 454 Life Sciences, Branford, Connecticut, USA)测序。所有非酒精性脂肪性肝炎(NASH)患者通过肝活检均符合Kleiner关于肝脂肪浸润、炎症和纤维化的标准。肥胖组招募了体重指数(BMI)高于第95百分位且肝功能测试正常的患者。健康对照组是BMI低于第85百分位的志愿者。此外,通过DINE健康饮食摄入量表(7.0.1版)和疾病控制中心(CDC)的食物频率问卷对所有个体的饮食摄入量(蛋白质、脂肪和碳水化合物)进行了评估。与此同时,该队列中的所有志愿者都被禁止饮酒,以确保酒精摄入量不会成为本研究中的一个混杂因素。
验证队列
本研究的外部验证是我们之前发表的一个独立队列。原始测序数据可通过EMBL-EBI(https://www.ebi.ac.uk/ena/data/view/PRJEB28350)获取。该队列包括31名健康对照组、14例无晚期纤维化的非酒精性脂肪肝患者和24例伴有肝硬化的NAFLD患者,并使用Illumina MiSeq进行测序。
宏基因组分析
在对16S rRNA基因测序数据进行质量控制后,以97%的序列一致性从头聚类获取微生物操作分类单元(OTUs),并通过Qiime2去除嵌合体。基于Silva-132-99参考序列,采用基于朴素贝叶斯分类器的物种分类器对微生物进行分类。不能精确注释到任何物种的OTUs通过NCBI Blast重新分配到同一属(或科)中序列最相似的菌种。最后,去除物种分类未分配的OTUs,进行后续分析。从发现队列和验证队列中分别获得样本覆盖率大于0.5的97个和190个OTUs。
微生物差异分析和样本分辨能力评估
采用Wilcoxon秩和检验来识别不同组间具有差异丰度的微生物,并采用Benjamini-Hochberg法来控制多重比较中的错误发现率(FDR)(FDR≤0.05)。利用受试者工作特征曲线下的面积(AUC)评估差异微生物的样本分辨能力。
基石菌种鉴定
该分析流程主要包括两个步骤:(1)采用基于因果推理的方法构建微生物因果相互作用网络;(2)采用基于微生物相互作用的动态干预模拟(DIS)确定基石菌种。
(1)通过因果推理构建相互作用网络
因果推理需要先验知识。考虑到微生物测序数据的组成特征,我们使用SparCC计算了肠道微生物组中菌种的共现情况。SparCC避免了相关分析中由于相对丰度造成的不准确性。先验网络中只包含与P ≤ 0.01的关系。
因果推理整合了两个主流的因果推理框架,Robins的潜在结果模型和Pearl的图形模型。我们的方法有效地控制了混杂因素的影响,并实现基于横断面数据的微生物因果关系的更准确推断。我们将因果推理的应用扩展到著名的微生物动态模型,generalized Lotka-Volterra模型(gLV)中。同时,采用迭代优化策略,提高了因果关系的准确性,并采用随机置换检验来确定显著性(P ≤ 0.01)。
(2)基石菌种鉴定
根据定义,基石菌种是维持或破坏微生物群落的完整性和稳定性的核心驱动因素。因此,微生物群的基石菌种能够作为疾病的潜在干预靶点。在此,基于微生物相互作用,我们应用DIS评估了每个候选基石菌种对整个肠道菌群的影响,从而确定对肠道菌群影响最大的基石菌种。
一个菌种的拓扑重要性是评价其对整个生态系统中其他菌种影响的关键指标,因此拓扑重要性可以作为基石菌种识别的重要依据。这里使用超链接引导主题搜索(HITS)网络节点重要性评估算法来量化群落中每个菌种的重要性。采用1000次随机置换检验来评估HITS评分的显著性。HITS得分显著较高的菌种(P ≤ 0.01)被定义为枢纽菌种。
参考前期研究,我们采用gLV方程构建了DIS中微生物相互作用和丰度变化的动态模型。DIS用于评估每个候选基石菌种干预后,菌群从患病状态中恢复至正常状态的能力。通过整合正常状态菌种的HITS得分来加权每个候选菌种的干预表现,用以奖励优先恢复正常微生物网络中具有更大拓扑重要性的菌种的干预策略。最后,使用迭代特征消除算法来确定组合干预得分最高的基石菌种。
微生物功能富集分析
利用KEGG数据库中的基因注释和模块的信息进行功能富集分析,以评估微生物的生物学功能。通过PICRUST2预测了微生物的同源信息。KEGG模块的富集分析采用单边Fisher精确检验,并采用Benjamini-Hochberg进行矫正。FDR ≤ 0.01的模块被认为是显著富集的模块。
统计分析
统计显著性由双侧Wilcoxon秩和检验、置换检验或单边Fisher精确检验确定。如果没有其他特别说明,本研究使用Python(3.6.0)进行统计分析。
代码和数据可用性
本研究没有新增的测序数据。处理后的数据已上传到NODE数据库(https://www.biosino.org/node/analysis/detail/OEZ013156)。本研究中使用的所有软件包都是开源的和公开的,本研究中使用的代码可以在GitHub的https://github.com/tjcadd2020/NAFLD_keystone上找到。所有的补充材料(文本、图、表、中文翻译版本或视频)也可从线上获取。
引文格式:
Wu, Dingfeng, Lei Liu, Na Jiao, Yida Zhang, Li Yang, Chuan Tian, Ping Lan, Lixin Zhu, Rohit Loomba, Ruixin Zhu. 2022. Targeting keystone species helps restore the dysbiosis of butyrate-producing bacteria in non-alcoholic fatty liver disease. iMeta. e61. https://doi.org/10.1002/imt2.61
-
点赞 (0人)
- 收藏 (0人)

