
iMeta | 扬州大学杜予州团队揭示同域内同食物的两种昆虫肠道微生物群落装配机制
- 看不见的线
- 3962
- 2024-06-07 09:16:08
- 文章来源:iMeta
同域内同食物的拟果蝇和黄粉鹿角花金龟肠道微生物组成主要受群落装配过程的驱动而非区域物种库

https://onlinelibrary.wiley.com/doi/10.1002/imt2.57
RESEARCH ARTICLE
●2022年10月13日,扬州大学植物保护学院杜予州团队在iMeta在线发表了题为“Gut microbiota composition in the sympatric and diet‐sharing Drosophila simulans and Dicranocephalus wallichii bowringi shaped largely by community assembly processes rather than regional species pool”的文章。
●本文发现同域内取食相同食物的拟果蝇与黄粉鹿角花金龟,两者肠道微生物的多样性、组成和网络结构存在显著差异,揭示了同域内的昆虫肠道微生物群落结构和功能的形成过程中,群落装配过程比区域物种库发挥更加重要的作用。
● 第一作者:朱玉溪
● 通讯作者:杜予州(yzdu@yzu.edu.cn)
● 合作作者:杨润、王欣宇、文涛、龚明辉、沈媛、徐珏烨、赵殿树
● 主要单位:扬州大学植物保护学院、南京农业大学资环学院、无锡市滨湖区农村农业局、佛罗里达大学
亮 点
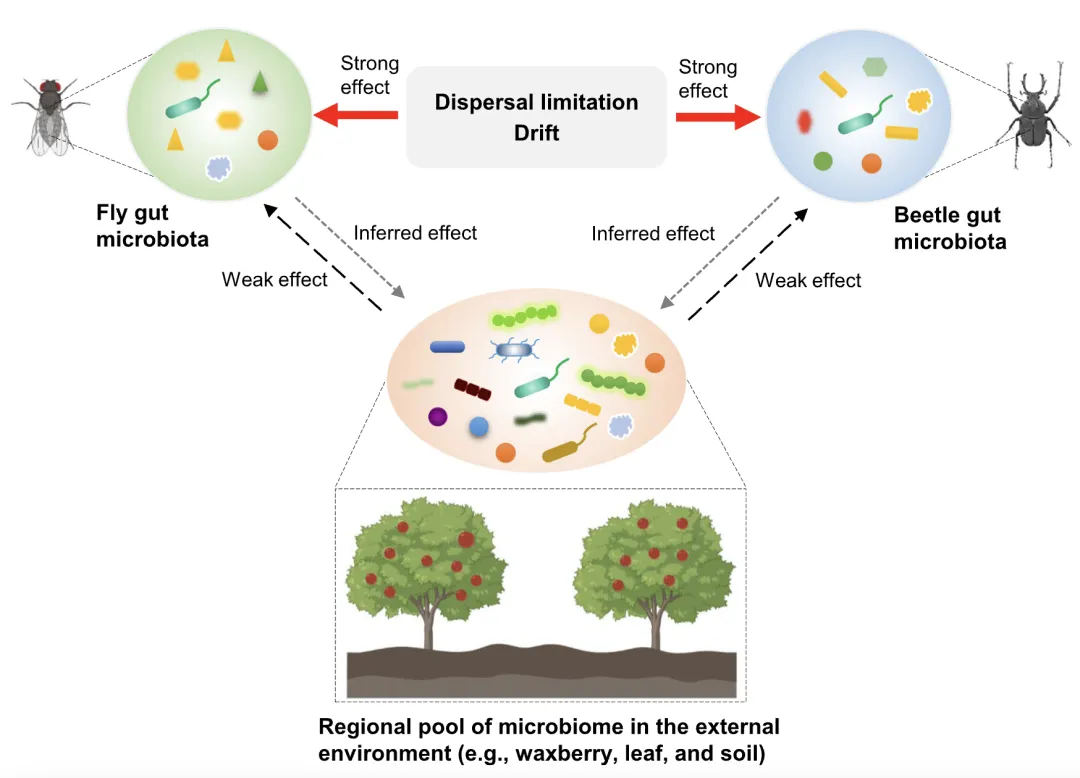
●同域内取食相同食物的拟果蝇与黄粉鹿角花金龟,两者肠道微生物的多样性、组成和网络结构存在显著差异;
●宿主种类通过改变群落装配过程的相对贡献塑造昆虫的细菌与真菌群落;
●拟果蝇与黄粉鹿角花金龟肠道微生物仅有少量来源于杨梅、树叶和土壤区域微生物库;
●昆虫肠道微生物的组成主要受装配过程的驱动而非区域物种库。
摘 要
解析个体从区域微生物库中的微生物群落装配机制是微生物生态学的核心问题,但在很大程度上仍未得到探索。本文研究了同域内取食相同食物(杨梅)的两种野生昆虫,即拟果蝇和黄粉鹿角花金龟的肠道细菌和真菌群落的装配过程和潜在来源。虽然在两种昆虫肠道中观察到一些趋同微生物,但两种昆虫肠道微生物的多样性、组成和网络结构存在显著差异。零模型分析表明,随机性过程(如漂移、扩散限制)在两种昆虫肠道真菌与细菌构建中发挥主要作用。然而,不同生态过程的相对贡献在两种昆虫肠道微生物装配中存在差异。此外,朔源分析表明,拟果蝇和黄粉鹿角花金龟肠道中只有少数微生物来源于杨梅、叶片或土壤等区域微生物库。功能预测分析暗示昆虫肠道菌群的物种特异性可能是由宿主功能需求和宿主-菌群协同进化特异性选择的结果。总之,我们的研究结果揭示了同域内的昆虫肠道微生物群落结构和功能的形成过程中,群落装配过程比区域物种库发挥更加重要的作用。
视频解读
Bilibili:https://www.bilibili.com/video/BV13K411D7ft/
Youtube:https://youtu.be/lY0HnUEYGUY
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
动物肠道中蕴藏着丰富多样的微生物。这些微生物在宿主的生理、生态和进化等方面发挥着至关重要的作用,例如影响宿主的适合度、消化、解毒、免疫、营养补偿以及参与宿主对逆境的响应等过程。尽管微生物对绝大多数动物至关重要,但有关昆虫肠道微生物的功能和多样性驱动因素在很大程度上尚不清楚。解析昆虫肠道微生物的塑造过程,对于靶向微生物的害虫绿色防控,及对有益昆虫的保护均具有重要的意义。
区域物种库是动物微生物群落的主要来源。动物体内的一些微生物类群,可在其食物或共享环境被追溯到;许多昆虫直接从其食物或周围环境中获取微生物。例如,蜜蜂通过从花丛环境中觅食来获取微生物。食物通常还可以快速、重复改变许多昆虫的肠道微生物群。一些物种如植食性毛毛虫,其肠道通常缺乏常驻微生物,它们从土壤而非寄主植物中获取微生物。综合以上结果表面,肠道微生物源于邻近的营养级或营养网络。
与此同时,中性理论强调了微生物群落的装配过程,特别是随机性过程在驱动微生物群落形成中的关键作用。通常来说,确定性和随机性过程协同调控微生物群落的装配,但这些生态过程(例如多样化、选择、扩散和漂移)在塑造宿主微生物群落中的相对贡献可能因系统而异。例如,最近对蜜蜂肠道微生物群的研究表明,漂移等随机性过程是蜜蜂肠道微生物装配的关键因素。相反,一些研究表明选择等确定性过程对宿主群落装配的影响更大。除此之外,这些生态过程的重要性也与宿主物种、食物或环境等因此密切相关。
因此,一个开放的核心问题是:区域微生物库和群落生态过程,哪个在塑造微生物群中占主导地位。在自然环境中,许多因素之间的复杂相互作用限制了这个问题的解决。为应对这一挑战,我们探索了两种不同昆虫物种即拟果蝇和黄粉鹿角花金龟的肠道细菌和真菌群落,它们有着共同的食物和栖息地。拟果蝇和黄粉鹿角花金龟的成虫分别使用舐吸式口器和咀嚼式口器取食成熟的水果,其中包括杨梅。尽管前期对拟果蝇和花金龟的微生物群落的研究已有报道,但关于这些昆虫自然种群个体肠道微生物群的来源和装配机制知之甚少。为了解决这一问题,本文对这两类昆虫肠道微生物群进行朔源分析及群落装配机制进行深入探究。我们首先调查不同类型样本间的微生物群落相似性,并探明昆虫肠道微生物类群是否来源于它们的食物(即杨梅)或环境(叶片或土壤)。其次,我们研究了两种昆虫肠道微生物群落的装配机制。最后,我们讨论了这两种昆虫肠道微生物的潜在功能。
结 果
不同样本间微生物的多样性、组成和网络结构
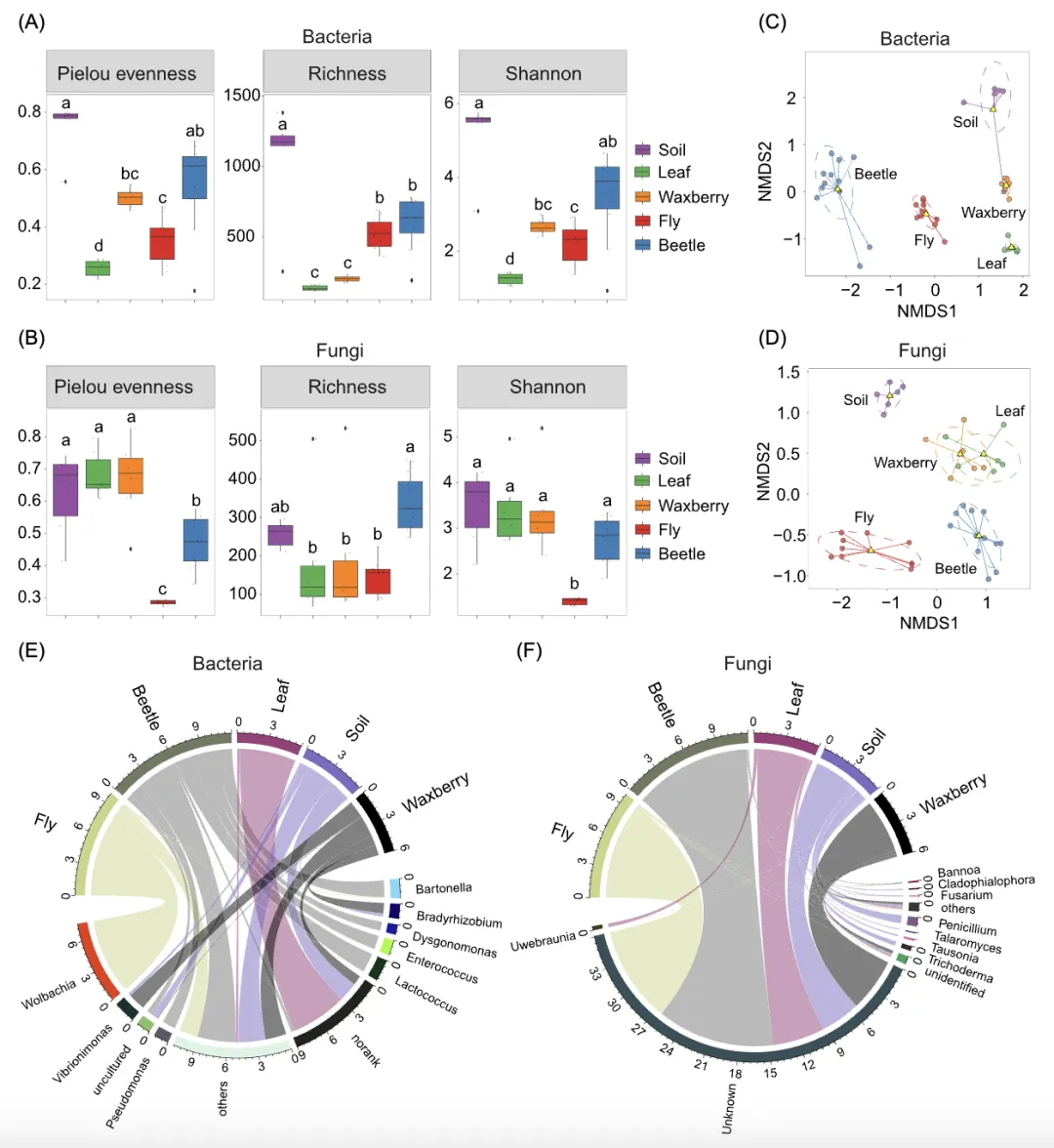
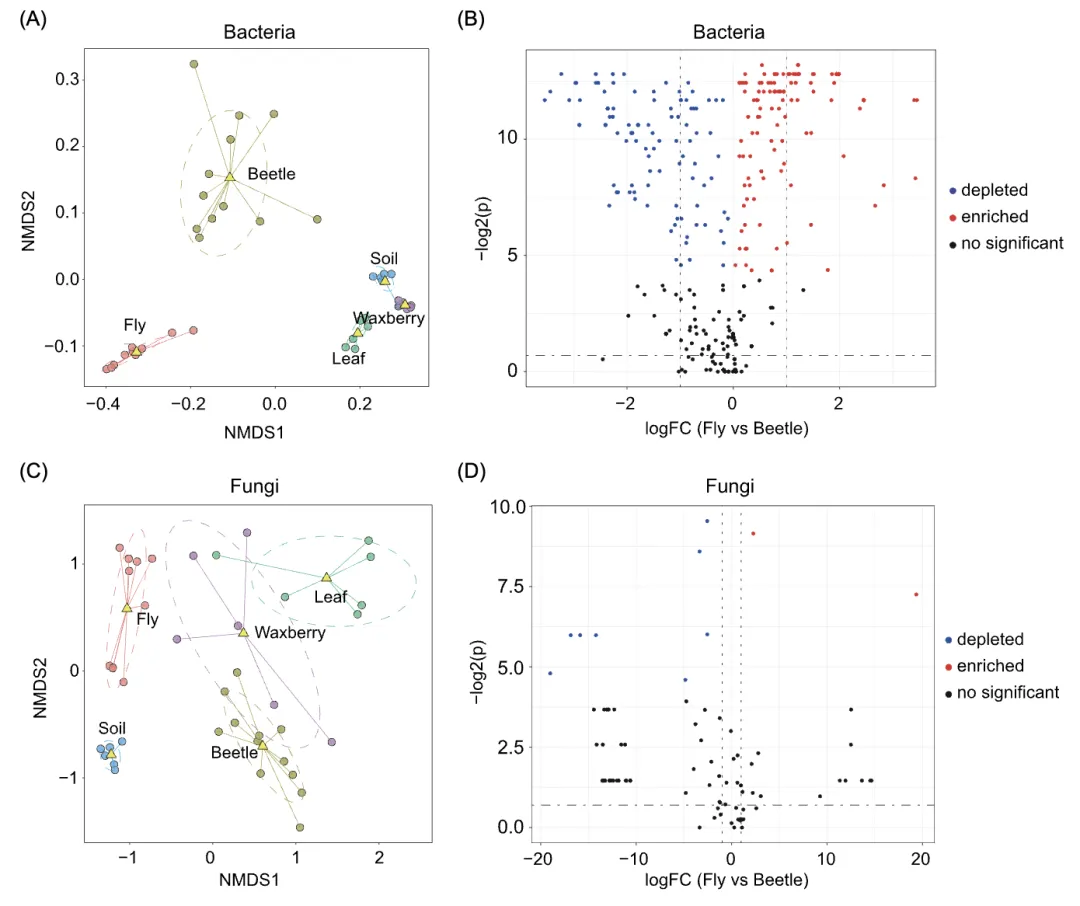
土壤、叶片、杨梅、拟果蝇和花金龟肠道间细菌与真菌群落的香农(Shannon)多样性指数均存在显著差异(图1A)。值得注意的是,在两种昆虫中,肠道细菌的群落多样性受宿主种类(F = 11.25,p = 0.004)而非性别(F = 3.13,p = 0.09)的显著影响,而肠道真菌的群落多样性受宿主种类(F = 114.90,p < 0.0001)和宿主性别(F = 6.66,p = 0.02)显著影响(表S1)。然而,与花金龟相比,拟果蝇肠道中细菌和真菌微生物的多样性均显著较低(细菌:p < 0.01;真菌:p < 0.0001)(图1A)。
细菌和真菌群落的组成在不同类型样品(土壤、叶片、杨梅、拟果蝇肠道和花金龟肠道)间差异显著(PERMANOVA,细菌:MRPP = 0.40,p < 0.001;真菌:MRPP = 0.55,p < 0.001;图1C、D)。果蝇肠道优势细菌为变形菌门Proteobacteria,而花金龟肠道的优势细菌属于厚壁菌门Firmicutes(图S1A)。在真菌方面,拟果蝇和花金龟的肠道几乎完全由未分类的菌群组成(图S1B)。在属分类水平上,细菌沃尔巴克氏体属Wolbachia、醋杆菌属Acetobacter、Commensalibacter和真菌镰刀菌Fusarium、Naganishia和青霉属Penicillium在拟果蝇肠道中具有较高的丰度。相比之下,在花金龟肠道中,乳球菌属Lactococcus、魏斯氏菌属Weissella、巴尔通氏体菌Bartonella、假单胞菌属Pseudomonas、肠球菌Enterococcus等细菌,以及Bannoa、Kwoniella、Hasegawazyma等真菌丰度较高(p < 0.05;图1E、F和图S2)。
网络分析表明,叶片(平均度Average degree:细菌:32.90;真菌:144.55)和杨梅(细菌:26.73;真菌:105.44)的细菌和真菌共生网络复杂程度明显高于拟果蝇(细菌:5.39;真菌:93.20)和花金龟肠道微生物(细菌:7.06;真菌:7.27)(图S3和表S2)。同样,叶片(细菌:6811;真菌:35197)和杨梅(细菌:1568;真菌:25781)的细菌和真菌群落的连接数多于拟果蝇(细菌:833;真菌:22367)或花金龟(细菌:1568;真菌:1531)(图S3和表S2)。花金龟肠道细菌网络比拟果蝇肠道细菌网络更复杂,但真菌网络则恰恰相反
每种昆虫体内细菌-真菌互作网络
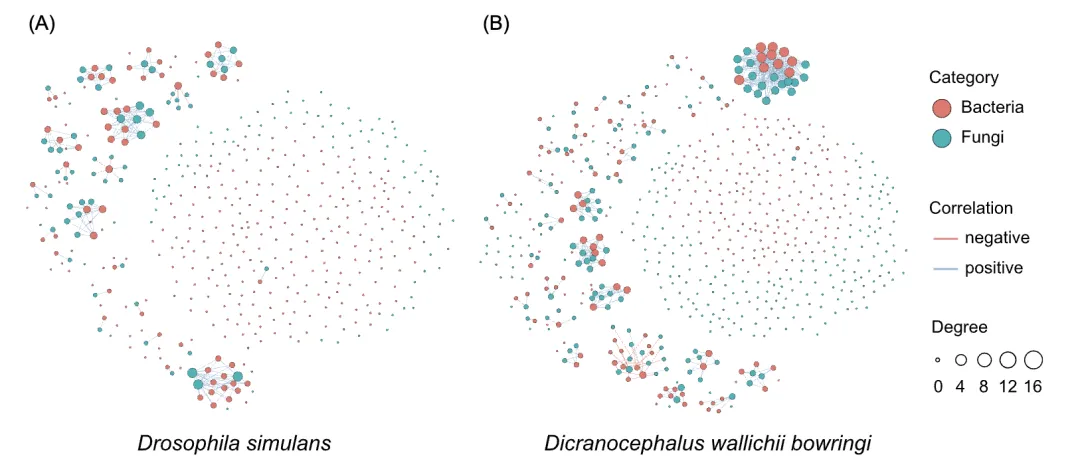
我们分别调查了两种昆虫的肠道细菌和真菌之间的相互作用。黄粉鹿角花金龟(Shannon:r = -0.51,p = 0.13;Richness:r = -0.47,p = 0.17)和拟果蝇(Shannon:r = -0.33,p = 0.30;Richness:r = -0.48,p = 0.12)体内的细菌和真菌之间的α多样性均无显著相关性,表明细菌和真菌在宿主体内可能具有不同的生态位。然而,网络分析表明,在同一昆虫宿主内,部分细菌与真菌密切相关(图2和表S3)。细菌和真菌之间的连接数,花金龟(366)高于拟果蝇(185)。同样,花金龟中的细菌和真菌网络的平均度高于拟果蝇(3.83 vs 3.25)(图2和表S4)。这些结果表明,在花金龟或拟果蝇中,部分细菌可能与真菌间存在互利共生关系。
土壤、叶片、杨梅和两种昆虫共有的微生物
在所有样品中,共获得4787个细菌OTUs和3394个真菌OTUs。土壤中含有细菌OTUs数目最多(n = 2258),其次是花金龟肠道(n = 1827)和拟果蝇肠道(n = 1766)(图S5)。相比之下,花金龟肠道含有最多的真菌OTUs数目,其次是拟果蝇肠道(图S5)。有105个细菌OTUs和25个真菌OTUs在花金龟和拟果蝇肠道中共同存在(图S5)。微生物溯源分析显示,拟果蝇和花金龟肠道细菌仅有1.98%和0.38%来源于杨梅细菌群落;而拟果蝇和花金龟肠道真菌分别只有0.16%和2.40%来源于杨梅真菌群落(图3)。在拟果蝇和黄粉鹿角花金龟肠道中,大部分的细菌和真菌都不是来自它们的食物(杨梅)或周围环境微生物群(叶片或土壤)(拟果蝇:细菌:97.52%,真菌:99.32%;花金龟:细菌:99.64%,真菌:95.88%;图3)。

果蝇与花金龟肠道细菌与真菌的群落装配
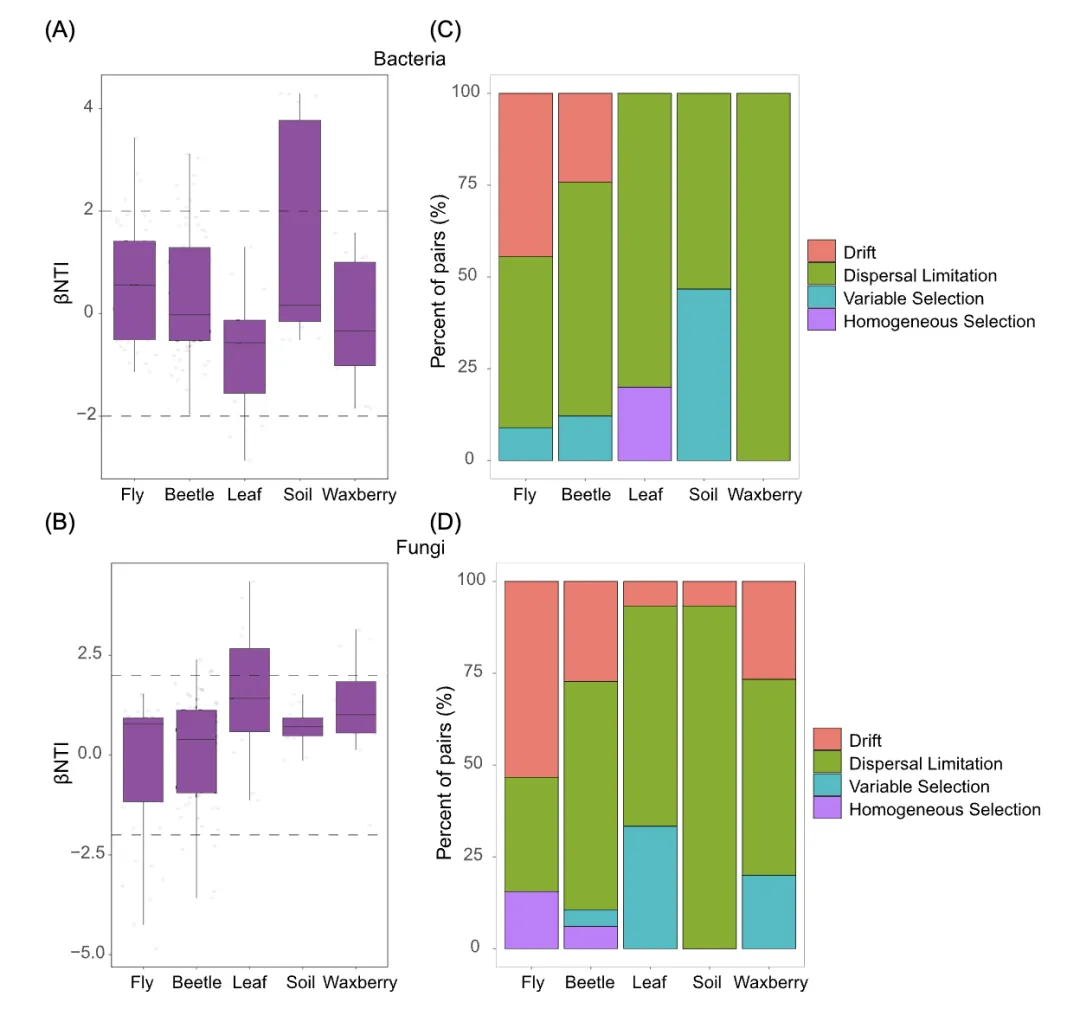
利用零模型分析量化了不同生态过程对两种昆虫微生物群落装配的相对贡献。结果表明,所有不同类型样本的细菌和真菌群落装配主要由随机性过程驱动(-2 < βNTI < 2)(图4A)。在拟果蝇和黄粉鹿角花金龟中,扩散和漂移是驱动2种昆虫肠道细菌和真菌群落构建的主要因子,其次,可变选择和同质选择分别在细菌群落和真菌群落中也起到一定作用(图4B)。然而,漂移对拟果蝇肠道细菌和真菌群落装配中的相对贡献(细菌:44.44%;真菌:53.33%)高于花金龟(细菌:24.24%,真菌:27.27%)。此外,扩散限制对花金龟微生物群落(细菌:63.64%;真菌:62.12%)的相对影响高于拟果蝇(细菌:46.67%;真菌:31.11%)(图4B)。总之,这些结果表明包括漂移和扩散的随机性过程,是驱动2中昆虫肠道细菌和真菌群落装配的关键因子,它们的相对贡献取决于宿主类别。
果蝇与花金龟肠道细菌与真菌的功能预测
鉴定出的细菌富含与代谢途径、碳代谢、脂肪酸代谢、肽聚糖生物合成、柠檬酸盐循环、戊糖磷酸化途径和双组分系统相关的基因(图S6A)。大量细菌基因的功能在拟果蝇肠道和花金龟肠道之间显著差异(Pair Adonis,MRPP = 0.071,p < 0.001)(图5A)。具体而言,拟果蝇和花金龟肠道细菌在ABC转运蛋白、磷酸转移酶系统、氨基酸代谢和嘌呤代谢等功能方面存在显著差异(图5B和表S5)。
基于FUNGulid预测分析,2种昆虫肠道真菌群落主要功能被归类为Pathotroph,Saprotroph和Symbiotroph(图S6B)。拟果蝇和花金龟肠道真菌中,在Saprotrophy,plant pathogens,fungal parasites和animal pathogens等功能间存在显著差异(Pair Adonis,MRPP = 0.499,p < 0.001)(图5C、D和表S5)。这些结果表明,肠道微生物群可能为2种昆虫宿主提供多样且特定的功能。
讨 论
理论上,个体肠道微生物的类群被认为是从区域微生物池中定殖的本地群落。然而,宿主肠道微生物的定殖往往受到一系列微生物群落装配过程的限制。这些潜在因素,包括区域微生物库和群落装配过程,在形成昆虫肠道微生物群落组成方面的相对重要性仍存在很大争议。在本研究中,我们发现生活在同一地区并且取食相同食物的两个不同昆虫物种之间,它们的肠道细菌和真菌群落多样性、组成和网络存在显著区别,这表明宿主类别强烈影响其肠道微生物群落。有趣的是,两种昆虫之间微生物群落的变化,可能是由群落装配过程的变化驱动,而区域环境微生物库(即食物、周围植物和土壤)影响较小。
我们发现拟果蝇和黄粉鹿角花金龟中只有极少数的肠道细菌和真菌类群可能获取于昆虫环境区域微生物库(杨梅、叶片和土壤),表明区域微生物库在2种昆虫肠道微生物群落塑造中的作用很弱(图6)。这也意味着,试图通过改变区域微生物库来打破昆虫肠道微生物群落平衡来进行害虫防控,这种害虫防控方法可能效果有限。此外,最近的一项研究表明,地上植食性昆虫重塑了寄主植物叶片微生物群落。这就提出了一个有趣的问题,即这2种昆虫是否也会改变其区域微生物库。多重生态位网络之间可能存在的复杂相互作用需要进一步研究。
之前有关野生拟果蝇和花金龟的研究表明,肠道细菌和真菌群落与宿主食物和栖息地密切相关。这意味着区域库,特别是食物,是许多昆虫获取微生物群的主要机制。利用实验室毛虫进行可控试验发现,植食性昆虫可能通过“搭便车方式”,直接从其食物或周围环境(例如土壤)中获取多种微生物。我们在2个不同类别昆虫(一个双翅目和一个鞘翅目)中的发现,与这些前期研究结果不同,尽管共享食物,这2种昆虫肠道中细菌和真菌群落组成仍呈现很大差异,因此我们认为这2种昆虫的肠道微生物具有宿主特异性。在更广泛的范围内,我们的结果支持之前的一些结论,即宿主种类比环境因素(例如食物)在塑造微生物的多样性和组成中发挥更核心的作用。因此,我们推测,共享的食物可能不足以干扰肠道微生物类群中物种特异性的差异。然而,在自然环境中,我们不能完全排除这些昆虫在转移为害杨梅之前是否取食其他水果的可能性。
多重的机制可能解释昆虫种类在驱动宿主肠道微生物群落的主导因子。首先,昆虫肠道具有独特的形态和生理条件,作为一个“过滤器”,导致许多来自环境库中的微生物无法在宿主肠道内定殖。这2种昆虫肠道的大小、形态和生理的不同,可能导致其肠道微生物组成的差异。其次,肠道共存的微生物可能会争夺有限的资源和空间。微生物间竞争格局差异可能导致不同昆虫肠道微生物群落的差异。在本研究中,我们还观察到同一宿主肠道中细菌-真菌互作。特别的是,影响黑腹果蝇肠道菌群结构的可遗传内共生体(例如沃尔巴克氏体),在拟果蝇肠道中也保持较高丰度。因此,我们推测微生物之间的相互作用是改变群落多样性和组成的重要因素。不管怎么样,这种可能性仍需进一步研究验证。最后,从生态学的角度来看,独立于宿主物种和性状的中性过程,以及不同物种间引起的微生物的装配过程差异,均导致不同昆虫物种之间微生物群落组成的差异。
我们发现漂移和扩散限制等随机性过程对拟果蝇和黄粉鹿角花金龟肠道内细菌群落装配具有重要影响(图6)。在斑马鱼肠道微生物群落的研究中,观察到类似结果,同样说明了随机性驱动因子在肠道微生物群落装配中的重要性。另一方面,一些其他昆虫肠道微生物群落装配的研究,强调选择等确定性过程在塑造群落结构中发挥着更大的作用。这些截然不同的研究结果表明,不同生态过程的相对贡献与宿主类别密切相关。近期的研究还表明,群落装配与地理变量等环境因素有关。在我们的研究中,漂移和扩散限制的相对作用在2种共享相同环境的昆虫肠道微生物塑造中不同。扩散限制导致微生物群落偏移,而漂移分散微生物群落。在一定程度上,这解释了为什么两个物种之间的肠道微生物群网络和组成可能存在很大差异。有关蜜蜂肠道细菌群落装配的研究推测:从长期共同进化的角度来看,确定性过程往往决定宿主与微生物共同进化的方向,而随机性过程是宿主与微生物共同进化的驱动力。在大多数情况下,这种宿主与微生物的共同进化倾向于互利共赢的共生模式。
大多数情况下,尽管肠道微生物群在分类和功能上存在多样性,但肠道微生物在不同宿主中起着比较保守的共生作用,主要参与宿主的新陈代谢、繁殖、病原体抵抗和免疫。在膜翅目、双翅目和半翅目在内的多种昆虫中,肠道微生物在宿主的生理功能中发挥着关键作用,如参与宿主对植物多糖消化和病原体防御。因此,了解肠道微生物群的功能,为通过打破共生控制害虫或保护经济昆虫健康提供了候选分子靶点。考虑到拟果蝇和黄粉鹿角花金龟在特定的生长发育阶段,需要以杨梅为食补充糖、氨基酸和其他必需营养元素,这一定程度上解释了为什么大多数肠道细菌都参与碳和氨基酸代谢。同样也提出了一个可能的问题,即宿主的特异性肠道微生物群是否归因于宿主的功能需求和宿主-菌群协同进化的选择结果。然而,对宿主的生理影响而言,昆虫肠道中细菌-真菌相互作用的功能相关性仍然未知。这些微生物群落的功能冗余程度以及其如何影响肠道微生物多样性和生态位重叠也不清楚。
引人注目的是,拟果蝇肠道中优势细菌为沃尔巴克氏体,沃尔巴克氏体是昆虫和其他节肢动物中最为常见的一种共生细菌,该共生菌以其多种方式操纵宿主生殖而闻名。之前的研究还表明,沃尔巴克氏体可以影响宿主免疫力,影响宿主对环境的适应,改变宿主-植物相互作用,并影响其他生态和生理功能。一些沃尔巴克氏体菌株也被认为能够介导拟果蝇的繁殖、适应性和免疫反应。为进一步了解沃尔巴克氏体作为控制害虫的潜力,沃尔巴克氏体和拟果蝇之间的相互作用,未来需要在自然环境背景下进行深入探索。
结 论
总之,我们发现在同域内2种共享食物的昆虫之间,其肠道微生物具有高度物种特异性,虽然随机性过程在推动微生物群落装配方面发挥了重要作用,但昆虫肠道中的大多数肠道微生物群落不太可能从区域微生物库中随机获得。因此,我们认为肠道微生物多样性和功能可能与宿主种的依赖性的群落装配过程密切相关,而不是从区域微生物库中获取。我们还讨论了肠道微生物群在两种昆虫中的潜在功能,未来的一个重要目标就是确定核心肠道细菌和真菌在这2种昆虫中的具体功能。
方 法
样品采集
所有样本均于2021年6月采自无锡杨梅园。我们随机选择了同域内的6棵杨梅树。黄粉鹿角花金龟和拟果蝇成虫采自成熟的杨梅果实(图S7)。此外,收集了昆虫取食的杨梅果实和周围杨梅叶片。样品保存在100%乙醇中,-20℃保存。土壤样本采自相同的6棵杨梅树,同1树体,随机收集其周围4个样点土壤(每个样地10-30g FW),混合后作为每棵树的1个样本。
拟果蝇成虫和黄粉鹿角花金龟成虫个体使用100%乙醇洗涤,然后用蒸馏水清洗3次。在无菌条件下,将昆虫个体置于1×磷酸缓冲盐溶液(PBS)中,用灭菌的镊子解剖整个肠道,整个肠道用无菌水洗涤3次,去除肠道内容物。在DNA提取之前,昆虫个体的肠道分别置于40 μL H2O的无菌离心管,-20℃条件下储存。
DNA提取,ITS1-2和16S rRNA扩增子测序
昆虫肠道DNA提取使用DNeasy blood and tissue kit试剂盒(Qiagen),按照试剂盒说明书进行。叶片、杨梅和土壤样品DNA的提取使用DNeasy Plant or PowerSoil Kit试剂盒(Qiagen)。分别使用1%琼脂糖凝胶和紫外光谱光度计(Nanodrop 2000)检测样本DNA的质量和数量。
DNA提取完成后,使用16S rRNA和ITS1-2扩增子高通量测序技术,分别分析叶片、土壤、杨梅和两种昆虫(拟果蝇和黄粉鹿角花金龟)样品中细菌和真菌的群落组成。细菌16S rRNA基因的V3-V4区域扩增使用引物341F(5′‐CCTAYGGGRBGCASCAG‐3′)和806R(5′‐GGACTACNNGGGTATCTAAT‐3′),而真菌的ITS扩增使用引物ITS1/ITS2(ITS1F:5′‐CTTGGTCATTTAGAGGAAGTAA‐3′)和ITS2R(5′‐GCTGCGTTCTTCATCGATGC‐3′)。
使用ABI GeneAmp® 9700仪器进行聚合酶链式反应(PCR)扩增。PCR混合物包含4μL 5×FastPfu缓冲液、2μL dNTPs (2.5 mM),每种引物0.8μL(5 μM),0.4μL TransStart Fastpfu DNA聚合酶,10ng模板DNA,最后加入ddH2O,使总体积达到20μL。PCR扩展条件如下:首先,95℃预热5 min;95℃ 30s,55℃ 30 s,72℃ 45s,30个循环;最后,72℃ 10 min。从2%琼脂糖凝胶中提取目标扩增产物,并使用AxyPrep DNA Gel Extraction Kit 试剂盒(Axygen Biosciences)纯化。在测序之前,将每个样品的产物以等摩尔浓度混合。使用Truseq DNA PCR-Free文库制备试剂盒分别构建细菌和真菌2个测序文库。样本送于上海凌恩生物科技有限公司测序,测序采用Illumina MiSeq 2500平台。
原始序列数据的处理过程采用标准化流程及之前描述的方法。使用QIIME2对原始序列过滤和组装。经过质控和去除嵌合体后,获得2991546条16S rRNA V3-V4扩增子序列和2507157条ITS扩增子序列,分别用于分析细菌和真菌群落。细菌的OTUs使用RDP对97%相似水平的OTU代表序列进行分类学分析,对比数据库为SILVA数据库,而真菌OTUs聚类分析使用UCLUST算法进行,与UNITE数据库进行匹配,置信度为80%。稀释曲线表明群落覆盖接近饱和(图S8)。在进行微生物分析之前,不同样本的测序数据进行抽平
真菌和细菌群落分析
所有的数据分析均使用R(version 3.6.2,http://www.r-project.org/)。
· α-多样性分析
使用“vegan”R包计算3个α多样性指数:Pielou均匀度、丰富度(观察到的OTUs数量)和Shannon多样性指数。双因素方差分析检验昆虫种类或性别对微生物香浓多样性指数的显著性影响。采用非参数统计检验(Kruskal-Wallis检验)分析不同昆虫肠道微生物α多样性间的差异。为了检验拟果蝇或花金龟体内细菌和真菌α多样性指数之间的相关性,使用了皮尔森 r系数和简单线性回归。
· β-多样性分析
细菌和真菌群落分析使用“vegan”R包进行,使用“ggplot2”将结果可视化。基于Bray-Curtis距离的非度量多维尺度(NMDS)分析来评估样品之间群落组成的差异性,并评估其β多样性。为了比较不同类型样品间细菌和真菌微生物群落组成的差异,使用adonis函数进行多元方差分析(PERMANOVA)。
· 群落组成分析
在多个分类水平上,对两种昆虫肠道微生物的相对丰度进行了分析。用Mann-Whitney U检验2种昆虫肠道细菌16S rRNA或真菌ITS基因在属水平的显著性差异(p < 0.05)。
· 共现网络分析
为了探究每个样品的细菌和真菌群落中OTUs的相对丰度之间的显著关系,使用“SpiecEasi”R包构建细菌和真菌关联网络,并使用“ggClusterNet”进行结果可视化绘制。所有ρ > |0.6|和p < 0.05的相关关系被认为是显著性相关。使用“vegan”和“igraph”R包来评估各种网络拓扑结构参数,包括聚类系数、平均度和平均路径长度、网络直径和集中程度。此外,将该网络与用“igraph”R包生成的随机结果进行比较,以识别任何非随机模式。网络的复杂性以“平均度”为指标,平均度值越大,代表网络的复杂性越高。
· 微生物朔源分析
利用Fast expectation‐maximization microbial source tracking (FEAST) (https://github.com/cozygene/FEAST)对2种昆虫肠道内真菌和细菌进行朔源分析,数据分析使用“FEAST”包。FEAST是一种基于期望最大化的方法,用于计算个体微生物群落来源于潜在环境中的比例。
· 零模型分析
零模型分析用于量化各个生态过程的相对贡献,包括漂移、选择和扩散。使用Ghost树(https://github.com/JTFouquier/q2-ghost-tree)构建真菌系统发育树,用FastTree2构建细菌系统发育树。通过将观测到的βMNTD与βMNTD的零分布平均值(999个随机)进行比较,计算βNTI。使用“picante”R包对标准偏差进行标准化,|βNTI| ≥ 2表示确定性过程在微生物群落装配中起主导作用,|βNTI| < 2则表明随机性过程的影响更大。我们将βNTI与基于Bray‐Curtis的Raup–Crick指数(RCI)结合起来,以量化生态过程,估算均质选择(βNTI < -2)、变量选择(βNTI > 2)、均质扩散(RCI < 0.95和|βNTI| < 2)、扩散限制(RCI > 0.95和|βNTI| < 2)和漂移(|RCI| < 0.95和|βNTI| < 2)驱动微生物群落的装配。
· 微生物功能预测
使用Tax4fun预测昆虫肠道细菌的潜在功能。Mann-Whitney检验比较2种昆虫间不同KEGG途径的差异水平。使用FUNGuild预测每个真菌OUT的生态功能
代码和数据可用性
本文涉及的高通量测序原始数据已在NCBI(PRJNA822659)发布;数据分析、绘图等R代码已在GitHub (https://github.com/taowenmicro/zhu.et.al.2022.07)发布。
引文格式
Zhu, Yu‐Xi, Run Yang, Xin‐Yu Wang, Tao Wen, Ming‐Hui Gong, Yuan Shen, Jue‐Ye Xu, Dian‐Shu Zhao, and Yu‐Zhou Du. 2022. Gut microbiota composition in the sympatric and diet‐sharing Drosophila simulans and Dicranocephalus wallichii bowringi shaped largely by community assembly processes rather than regional species pool. iMeta. e57. https://doi.org/10.1002/imt2.57
-
点赞 (0人)
- 收藏 (0人)

