
微生物组研究中 OTU 的前生今世
- 看不见的线
- 7396
- 2024-07-04 17:25:59
- 文章来源:生信菜鸟团
本文翻译整理自 Robert Edgar 实验室官网:https://drive5.com/
定义和解释 OTUs
Sneath-Sokal OTUs
在更早的年代,早于 DNA 测序,早于系统发育树构建算法(例如 neighbor-joining 和 maximum likelihood),操作分类单元 (Operational Taxonomic Unit, OTU) 的概念就由 Peter Sneath 和 Robert Sokal 在 1960 年代通过一系列数值分类学领域的书籍和文章引入了(参见 Sneat & Sokal: Numerical Taxonomy, WH Freeman, 1973)。他们的目标是开发一种定量策略,根据观察到的特征将生物分类,创建层次分类,尽可能忠实地反映生物之间的进化关系。Sneath-Sokal OTUs 是以数值描述的观察性状表来构建的;例如1=存在,0=不存在。层次结构(树)是根据它们共同特征的数量反复合并最相似的组来进行构建。这是应用于生物分类的凝聚聚类算法的早期示例。
数值分类法是生物系统学中的一个分类系统,它根据分类单元的特征状态通过数值方法对其进行分组。它旨在使用诸如聚类分析之类的算法而不是对其属性进行主观评估来创建分类法。该概念最初由 Robert R. Sokal 和 Peter H. A. Sneath 在 1963 年提出。他们将该领域划分为基于整体相似性模式形成分类的表型学和基于分类群估计进化历史的分支模式进行分类的分支学。
曾经的 97% 同一性阈值
在 16S 测序中,OTU 通常使用 97% 的同一性阈值构建。第一次提到这个阈值是在 (Stackebrandt and Goebel 1994)。Stackebrandt 和 Goebel 发现 16S 序列的 97% 相似性大约对应于 70% 的 DNA reassociation value,而 DNA reassociation value 在这之前已被用于进行细菌物种的定义(Wayne 等,1987)。
现在的 OTU 同一性阈值
2014 年进行的一项分析发现,通过对全长 16S 序列进行聚类来逼近物种的最佳阈值为 98.5%(Yarza 等人,2014 年)。但这项研究使用了 CD-HIT 进行聚类分析,CD-HIT 对序列同一性的定义并不适用于作为进化距离的衡量标准。另一方面,这篇研究基于 SILVA 数据库中的分类进行注释,而这个数据库中有大约五分之一的注释可能是错误的(Edgar 2018a)。在最近的一篇研究表明,全长序列的最佳聚类阈值为 ~99% 的同一性,V4 的最佳聚类阈值为 ~100% (Edgar 2018b)。
CD-HIT 序列同一性
第 6 版及更高版本的 USEARCH 使用的序列同一性为 BLAST 定义,即相同碱基的数量除以对齐碱基的总数。早期版本的 USEARCH 支持多种定义,默认使用 CD-HIT 标识定义。CD-HIT 定义是:CD-HIT 同一性 = 相同碱基的数量 / 较短序列的长度
这个定义在实践中存在许多问题。对于给定给定两条序列,BLAST 同一性<= CD-HIT 同一性。这是因为 BLAST 会对 gaps 进行罚分,而 CD-HIT 有时不会(在极端情况下,可以和任意数量 gaps 对齐,从而导致 100% 同一性的极端值)。一般来说,插入和删除的可能性通常低于替换。因此,作为进化距离的衡量标准,gaps 应该与替代一样重要,所以 BLAST 定义在生物学上更现实。
CD-HIT 是定义相比于 BLAST 的来说,其对比对参数的选择更为敏感。这是因为缺少 gap 罚分往往会产生更高同一性的“gappier”比对。举个例子:
GATTTACATT
||| | ||
GATACATCTT根据 BLAST 和 CD-HIT,该比对的同一性都为 6/10 = 60%。现在,假设我们取消 gaps 罚分,那么下面的比对方式将导致他们有更高的序列同一性:
GATTTACA--TT
|| |||| ||
GA--TACATCTT现在 BLAST 的同一性是 8/12 = 67%,而 CD-HIT 的同一性是 8/10 = 80%。请注意,CD-HIT 同一性的变化更大。
CD-HIT 定义倾向于在给定的同一性阈值下产生较少的集群(更大的平均大小),但这些集群的质量往往较低。
用 16S 序列聚类构建 OTU
用于 16S 序列聚类的最早程序是 FastGroup(Seguritan 和 Rohwer 2001)。FastGroup 使用了类似于 UCLUST 的贪婪聚类算法,同一性阈值定义为 97%。DOTUR (Schloss and Handlesman 2005) 是 mothur (Schloss et al. 2009) 的前身,使用约束聚类和选项来使用最大、最小或平均链接。DOTUR 和 mothur 使用了相同的同一性阈值来生成 OTU,并指出(DOTUR 论文)“[s] 大于 97% 同一性的序列通常分配给同一物种,95% 同一性的序列通常分配给同一属,而 80% 同一性的序列通常被归入同一门,尽管这些区别是有争议的”。QIIME (Caporaso et al. 2010) 几乎完全使用 97% 的阈值。早期版本的 QIIME 使用 CD-HIT 作为默认的 OTU 聚类方法,后来被 UCLUST (Edgar 2013) 取代。
OTU 作为物种的操作性定义
当进入测序的时代,我们可以通过 PCR 和 Sanger 测序得到 16S 序列。那时,公共数据库中的 16S 序列还相对较少,因此大多数序列还无法通过数据库搜索识别。一种实用的替代方法是使集群具有 97% 的同一性。这些集群可以被视为 Sneath-Sokal OTU,暂时将序列分配给物种或更高级别的组。如果但这仅限于序列中的实验误差比较小的时候。在早期的文献中,很少(从不?)考虑这些测序或实验错误。这可能是因为 Sanger 测序的错误率非常低,另一方面,PCR 引入的错误,尤其是嵌合体,在 90 年代并未广泛为人所知(Ashelford 等人,2005 年)。
基于 NGS reads 的 OTU
下一代测序 (NGS) 出现在 1990 年代后期,通过对 16S 基因的低成本、高通量测序,彻底改变了微生物学。与 Sanger 测序相比,这些优势也伴随着更短的读长和更高的错误率。OTU 聚类方法也开始应用于 NGS reads,但人们逐渐清楚,由于实验误差,这些方法产生了许多虚假的 OTU,导致对多样性的估计过高(参见例如 Huse 等人,2010 年)。在撰写本文时(2017 年年中),一些广泛使用的方法也在持续产生大量的虚假 OTU,特别是 QIIME。
Westcott 和 Schloss OTU
在 Westcott and Schloss 2017 中,Westcott 和 Schloss 基于 Matthews 相关系数 (MCC) 提出了 OTU 的定义。但是,它们的定义在实践中仍存在一些问题。
具体定义如下:
1.同一 OTU 中的所有序列配对必须至少有 97% 的相似度。
2.不同 OTU 中的所有序列配对的相似度必须小于 97%。
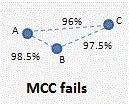
然而,如一个简单的例子所示,使用真实数据通常无法满足这些要求。
假设有三个菌株 A、B 和 C,16S 相似度 AB=99%、BC=98% 和 AC=96%,如下所示。
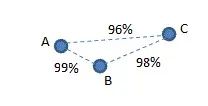
根据 Westcott 和 Schloss 的定义,A 和 B 应该属于同一个 OTU,而 B 和 C 应该属于同一个 OTU,因为这两个对具有 >97% 的同一性。理论上 A 和 C 也属于同一个 OTU,但根据定义,他们并不属于同一 OTU,因为 他们的相似性 <97%。因此,在此示例中,无法根据定义生成有效的 OTU。
理想中的 OTU
类物种组的操作定义
序列的 Sneath-Sokal 分类。这就是 16S OTU 诞生的历史动机。
将单个菌株内的变异合并为一个集群
细菌染色体通常包含多个拷贝的 16S 基因(旁系同源物)。旁系同源物之间的序列差异通常较小。聚类倾向于将旁系同源物合并为单个 OTU。如果每个表型都只对应一个 OTU,这对大多数分析都是非常有意义的。
将单一物种的菌株之间的变异合并为一个集群
对于给定种内的不同菌株,例如大肠杆菌,其 16S 序列通常存在一些差异。旁系同源物也会进一步增加种内的变异水平。聚类倾向于合并这两个级别的变异,为种创建单个 OTU 或至少更少的 OTU。如果目标是为每个表型获得一个 OTU,这不是一件好事,因为不同的菌株在表型上可能存在重要差异(例如,致病性与否)。此外,虽然菌株的概念相当明确,但种的概念对细菌来说仍有些模糊(Gevers 等人 2005,Doolittle 和 Pakpke 2006),因此每一菌种对应一条 OTU 可能并不是一件好事。
将实验误差引起的变异合并到一个集群中
PCR 引入的错误也会导致 reads 中的序列变异,这些变异并非源自生物学本身。聚类也会倾向于将错误序列与正确的生物序列合并。为了有效生成 OTU,我们会要求过滤 > 3% 错误率的序列,因为它们总是会产生虚假的 OTU(参见 Edgar 和 Flyvbjerg 2014)。即使是误差 < 3% 的序列也可能导致假的 OTU,这取决于相应序列是靠近集群中心还是靠近边缘(更有可能)。
UPARSE OTU
UPARSE-OTU 算法(cluster_otus命令)旨在报告正确生物序列的子集,以便 (a) 所有 OTU 配对的同一性 <97% ,以及 (b) OTU 是其邻域中丰度最高的序列。这些可以解释为 Sneath-Sokal OTU,给出物种的操作近似。
降噪 OTU (ZOTU)
使用 UNOISE 算法(unoise3命令),目标是报告 reads 中所有正确的生物序列。这些称为零半径 OTU 或 ZOTU。由于种内变异(旁系同源物之间的差异和菌株之间的差异),某些物种可能会被分成几个 ZOTU。ZOTU 的优势在于它们能够解析具有潜在不同表型的密切相关菌株,这些菌株可能会被 UPARSE-OTU 算法归为同一簇。
OTU 丰度
根据我们的定义,OTU 是序列,而不是簇。传统的 OTU 分析通常基于 OTU 表,这需要每个样本中每个 OTU 的丰度或频率。我们通过计数 reads 来计算丰度,同时强调 reads 丰度与物种丰度的相关性非常低,因此对这些丰度的生物学解释尚不清楚。
对于降噪序列,ZOTU 的丰度为源自该(唯一的)模板序列的 reads 数量,计算正确 reads 和错误 reads 。
对于 97% 聚类的 OTU (UPARSE),OTU 的丰度是指从所有生物序列中得出的 reads 数量,这些生物序列与 OTU 序列的同一性 >=97%,计算正确 reads 和错误 reads 。这个定义还不够,因为一条 read 可以匹配两个或多个 OTU。如果有多个选择,read 将分配给具有最高同一性(不是最高丰度)的 OTU,因为具有最高同一性的 OTU 更有可能属于同一物种。如果选择仍然不明确,则选择数据库文件顺序中的第一个 OTU。此过程可确保给定菌株的 reads 应始终分配给相同的 OTU。
实际上,对于 OTU 和 ZOTU,丰度是通过使用otutab命令以 97% 的同一性阈值将 reads 与 OTU 进行比对来计算的。将 reads 分配给最相似的 OTU 序列。在 97% OTU 的情况下,使用 97% 同一性阈值的动机是不言自明的。而对于 ZOTU,97% 的阈值表示我们可以容忍由于测序和 PCR 导致 3% 的错误。
关于 UPARSE 算法
UPARSE-OTU 算法基于 NGS 扩增子 reads 构建 OTU 代表序列。reads 需进行预处理,例如合并双端序列、去除 barcode、质量过滤和全局修剪。生成 OTU 后再进一步将 reads 映射到 OTU 并构建 OTU 表。
输入序列
UPARSE-OTU 的输入是一组序列。每个序列都标有一个整数值,表示其丰度。在实践中,丰度通常是具有给定唯一序列的 reads 的数量,但它也可以是降噪步骤后扩增子的预测丰度。
聚类标准
UPARSE-OTU 的目标是确定一组满足以下标准的 OTU 代表序列(输入序列的子集)。
1.所有配对 OTU 序列的成对序列同一性 <97%。
2.OTU 为 97% 邻域内丰度最高的序列。
3.嵌合体序列被丢弃。
4.所有非嵌合体输入序列应至少匹配一个 >= 97% 同一性的 OTU。
贪婪聚类
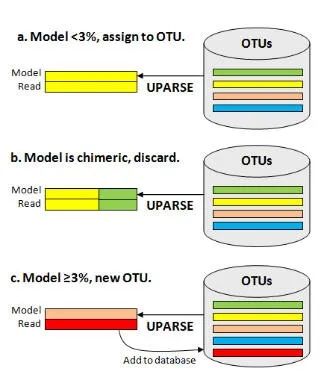
UPARSE-OTU 使用贪婪聚类算法来寻找生物学相关的解决方案,如下图所示。高丰度 reads 更有可能是正确的扩增子序列,因此也更有可能是真正的生物序列,这里 UPARSE-OTU 按丰度递减的顺序考虑所有的输入序列,OTU 质心倾向于从丰度更高的 reads 中选择。
每个输入序列与当前的 OTU 数据库进行比较,并使用 UPARSE-REF 找到该序列的最大简约模型(下图)。有以下三种情况。(a) UPARSE-REF 模型与现有 OTU 的序列同一性 >= 97% ,(b) 模型为嵌合体,或 (c) 模型与现有 OTU 同一性 <97%。在情况 (a) 中,输入序列成为 OTU 的成员。在情况(b)中,输入序列被丢弃。在情况(c)中,输入序列被添加到数据库中,成为新 OTU 的代表序列(质心)。
UPARSE-REF
给定一个样本的序列参考数据库 D,UPARSE-REF 使用简约推断序列中的错误。UPARSE-REF 的目标是用最少的事件从 D 中的序列推断给定的序列 S。这里,“事件”是指由 PCR 或测序错误引起的突变。我们通过使用来自数据库(refseqs)的一个或多个序列构建模型序列 M 来进行推断。通常,M 是表示非嵌合体扩增子的单个 refseq,少数情况下 M 由 m 个 refseq 片段组成,这些片段连接起来代表一个嵌合体扩增子。如果 M 为一个片段,即是单个 refseq,则 M 和 S 之间的距离定义为错配的碱基数量,这些错配被解释为测序或 PCR 错误。
下图显示了嵌合体模型 reads 的示例。这里,嵌合交叉的罚分是+3,错配的罚分是+1。该模型的总分是 4(每一个错配+1,每一个嵌合交叉+3)。
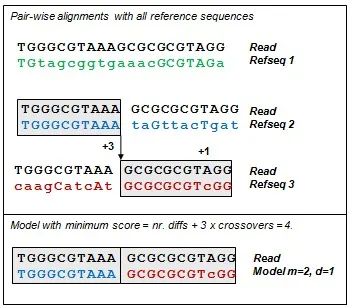
UPARSE-REF 用作 UPARSE-OTU 算法中 OTU 构建的步骤(cluster_otus命令)。UPARSE-REF 作为独立命令 (uparse_ref) 的主要用途是在模拟数据中注释 reads、OTU 或其他序列,这些样本中的生物序列集是已知的。
Reference
Edgar, R.C. (2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads,Nature Methods[Pubmed:23955772, dx.doi.org/10.1038/nmeth.2604].
使用 UPARSE 还是 UNOISE
OTU 有两种不同的生成方法:97% 聚类和降噪。
•UPARSE 算法生成 97% 的 OTU。
•UNOISE 算法进行降噪。
应该选择哪个?建议两个都试试。如果生物学结论不同,那么你应该担心这两个结果都不可信,并尝试了解为什么会发生这种情况。如果两种方法都一致,则往往会互相印证。(非要选一个,选择降噪算法)
UCLUST 算法
UCLUST 算法也是将一组序列聚类为集群。但 UCLUST 并不是为 OTU 聚类而设计的。
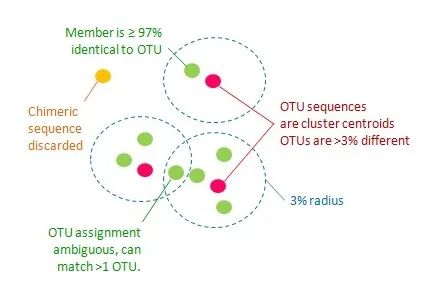
集群由一条序列进行定义,称为质心或代表性序列。集群中的每个序列与质心序列之间都大于给定同一性阈值,如下图所示。同一性阈值 (T) 可以视为集群的半径。聚类命令包括cluster_fast和cluster_smallmem。
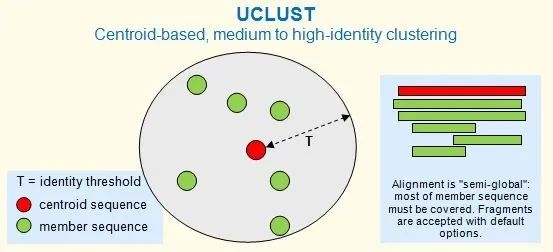
推荐的同一性范围
UCLUST 在蛋白质的~50% 及以上和核苷酸的 ~75% 及以上的同一性下有效。在较低的同一性下,这种类型的方法是有问题的,因为 (i) 比对质量下降和 (ii) 无法从比对中可靠地确定同源性。
聚类标准
UCLUST 旨在找到一组集群,使得:
1.所有质心彼此之间的相似度 < T,并且
2.所有成员序列与质心的相似度 >= T。
使用默认参数,该算法是启发式的,并且不能保证条件 (1) 成立,尽管在实践中假阴性(两个相似度 >= T 的质心)很少见。注意,通常,许多不同的聚类都满足这些标准。例如,一个序列可能匹配到两个不同的质心,其同一性都 > T。理想情况下,它会被分配到最近的质心,但仍有可能有两个或更多相同距离的质心,在这种情况下,最佳聚类分配是不明确的。
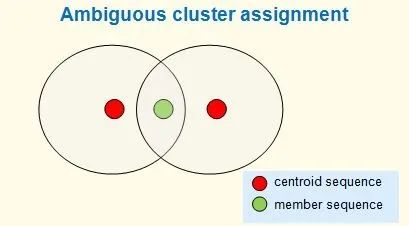
这里的同一性是使用全局比对来计算的。
输入序列顺序
UCLUST 是一种贪婪算法,输入序列的顺序很重要。在cluister_smallmem命令中,序列按照它们在输入文件中出现的顺序进行处理。如果下一个序列与现有的质心匹配,则将其分配给该集群,否则成为新集群的质心。这意味着我们需要先对序列进行排序,以便最合适的质心倾向于出现在文件中的较早位置。最佳顺序可以使用序列长度或者丰度。cluster_fast命令可以使用输入序列顺序(默认值),也可以分别指定 -sort length 或 -sort size 来按序列长度或按大小注释排序。
检索质心数据库
UCLUST 的一个基本步骤是将输入序列与目前识别的质心进行比较(这里使用 USEARCH 算法完成)。大多数 USEARCH 参数,包括索引选项,都可以与聚类命令一起使用,以控制敏感度、内存使用和速度。
-
点赞 (0人)
- 收藏 (0人)

