
统计FastQ/FastA文件信息
- 看不见的线
- 1185
- 2025-07-04 08:32:44
- 原创
Seqkit2是 Seqkit的新版本,是一个专门用于处理和分析生物序列数据的软件工具。它支持多种序列数据格式,包括FASTA、FASTQ等,并提供了一系列有用的功能,如数据处理、过滤、统计、格式转换等,是生物信息学领域中常用的工具之一。以下是使用seqkit统计FastQ/FastA文件信息。
stat/stats子命令:FASTA/Q文件的简单统计
# Usage: seqkit stats [flags]
# flags:
-N, --N strings:添加其他类似 N50的统计作为新列。值范围为 [0, 100],支持多个值,例如 -N 50,90或 -N 50 -N 90。
-a, --all:显示所有统计信息,包括序列长度的四分位数、总间隙数、N50等。
-b, --basename:只输出文件的基本名称,不包含路径信息。
-E, --fq-encoding string:指定 FASTQ质量编码格式。可用值为 'sanger'、'solexa'、'illumina-1.3+'、'illumina-1.5+'、'illumina-1.8+'。默认为 'sanger'。
-G, --gap-letters string:指定间隙字符,默认为 "- ."。
-h, --help:显示关于 stats子命令的帮助信息。
-e, --skip-err:跳过错误,仅显示警告消息。
-S, --skip-file-check:在给定文件或文件列表时跳过输入文件检查。
-i, --stdin-label string:替换标准输入的默认标签 "-"。
-T, --tabular:以机器友好的表格格式输出统计信息。
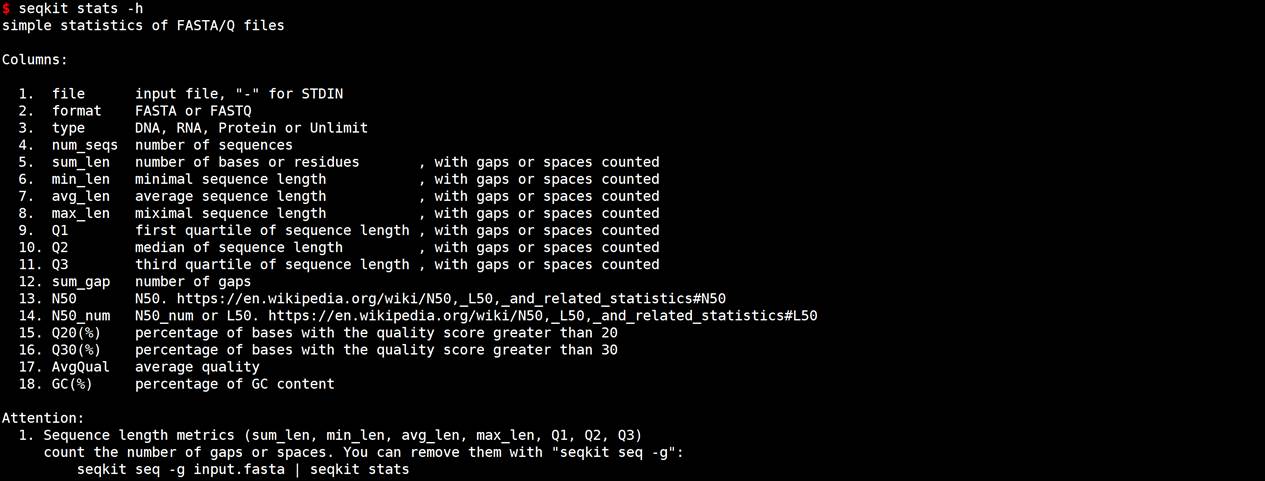
#帮助信息查看
seqkit stats -h
#下载参考序列,一个fastq文件,两个fasta文件
wget http://data.biostarhandbook.com/reads/duplicated-reads.fq.gz
wget ftp://ftp.ncbi.nih.gov/refseq/release/viral/viral.1.1.genomic.fna.gz
wget ftp://ftp.ncbi.nih.gov/refseq/release/viral/viral.1.protein.faa.gz
#统计FastQ/FastA文件信息
seqkit stat *.gz

#显示所有统计信息
seqkit stat -ab *.gz

#将统计结果保存为制表符分隔文件
seqkit stats -ab -T *.gz > stats_output.tsv


-
点赞 (0人)
- 收藏 (0人)

