
干货满满丨小白の名词解释小课堂——扩增子相关①
- 看不见的线
- 5800
- 2024-07-04 17:08:59
- 文章来源:凌波微课
第一堂课,让我们来了解一下扩增子测序的基础知识、α多样性。
微生物多样性测序,又名扩增子测序,是对特定长度的PCR产物或者捕获的片段进行测序,主要包括16S rRNA测序、18S rRNA测序、ITS测序及目标区域扩增子测序等。采用高通量测序平台测定的16S/18S /ITS全长或者某个高变区域的序列,来反映环境样品在细菌、真菌、古菌分类方面物种之间的差异,对研究海洋、土壤、肠道粪便等环境中的微生物构成有重要的指导作用。
基础知识部分
16S rRNA
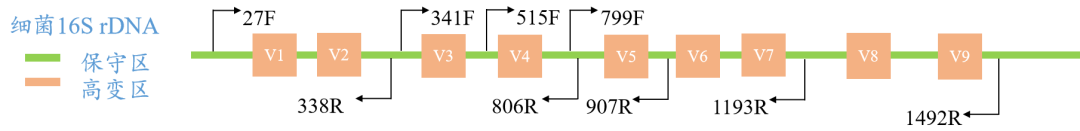
:16S核糖体RNA(16S ribosomal RNA),简称16S rRNA,是原核生物的核糖体中30S亚基的组成部分。16S rRNA的长度约为1,542 nt,具有高度的保守性和特异性。物种间16S rRNA序列既有高变区(V区,物种之间有差异)也有保守区(物种之间高度相似),呈交替排列,原核16S rRNA序列包含9个高变区,保守区反映了物种间的亲缘关系,而可变区则反映了物种间的差异。这两个区域呈交替排列,保守区可用于设计通用引物进行目的片段的扩增,通过对高变区的分析可以辨别细菌种类。(注:16S菌群测序是以DNA为研究对象,而非RNA。)
16S rDNA全长测序:通过PCR的方法扩增目标样品微生物群落16S rDNA的全长,利用三代测序技术长读长的特点,可以得到微生物群落16S rDNA的完整信息,从而提供比二代测序更为准确的物种分类学注释结果,进而比较不同样品间微生物群落的差异。
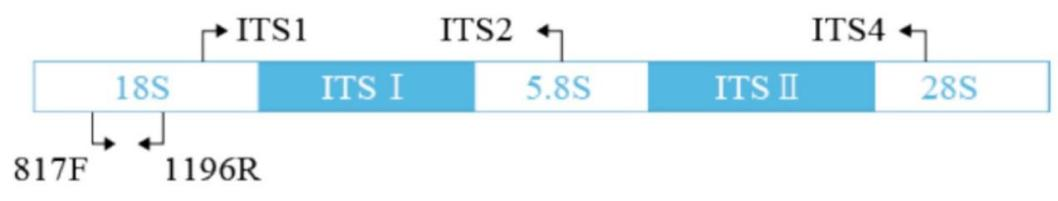
ITS测序:该类测序主要对环境微生物中的真菌多样性进行分析。ITS(InternalTranscribed Spacer)是内转录间隔区,是真菌核糖体RNA(rRNA)基因非转录区的一部分,位于真菌18S、5.8S和28S rRNA基因之间,分别为ITS 1和ITS 2。用于真菌鉴定的ITS序列通常包括ITS1、5.8S 和ITS2。真菌ITS 区域长度一般在500~750bp。在真菌中,5.8S、18S和28S rRNA基因具有较高的保守性,而ITS由于承受较小的自然选择压力,在进化过程中能够容忍更多的变异,在绝大多数真核生物中表现出极为广泛的序列多态性。同时,ITS的保守型表现为种内相对一致,种间差异较明显,能够反映出种属间,甚至菌株间的差异。此外ITS 序列片段较小、易于分析,目前已被广泛应用于真菌属内不同种间或近似属间的系统发育研究中。
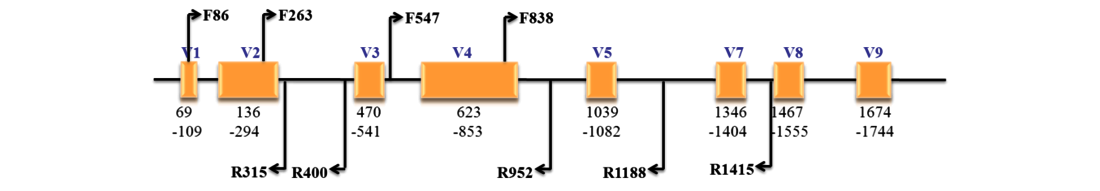
18S rRNA测序:18S rRNA为编码真核生物核糖体小亚基rRNA的DNA序列,反映样品中真核生物之间的种类差异。与细菌多样性分析类似,在真核微生物中也有三类核糖体RNA(rRNA),包括5.8S rRNA、18S rRNA和28S rRNA。18S rRNA基因是编码真核生物核糖体小亚基的DNA序列,其中既有保守区,也有可变区(V1-V9,没有V6区)。保守区域反映了生物物种间的亲缘关系,而可变区则能体现物种间的差异,适用于作种级及以上的分类标准。
功能基因:功能微生物是在自然界中由于其功能的重要性而受到广泛关注的一类微生物,如硝化细菌、反硝化菌、氨氧化细菌、硫酸盐还原菌、固氮菌等。每种功能微生物在分类学上可能有很大不同,但却具有相类似的基因使其能够发挥同样的功能,因此使这些功能细菌发挥这种特定功能的基因就称为功能基因,如nxrA、nirS/nirK、amoA、dsrB、nifH、nifH。功能基因测序一种针对功能基因目标片段进行扩增测序的分析技术。借助合适的类群特异性引物,可以检测菌群中特定种类/功能微生物的特征序列,反映菌群中该特定类群微生物的组成结构,从而发现它们的分布特征,阐明样本间多样性和组成差异,进而揭示该类群微生物中与差异相关联的关键成员。
OTU:可操作分类单元 (OTU)。为了便于进行分析,人为给某一个分类单元设置的统一标志,在16S rDNA微生物多样性研究中,要了解一个样品测序结果中菌种、菌属等数目信息,需要将测序得到的Tags按照碱基相似度97%进行聚类,得到的每一个聚类称为一个OTU,普遍认为相似度97%可以区分属水平的不同微生物,之后在每个OTU中选取一条代表序列进行物种分类学注释,也就是说每个OTU会对应一个物种信息。
ASV:传统的OTU聚类的步骤实际上会掩盖部分存在测序错误的序列,导致部分OTU的丰度出现不准确,同时序列真实的变异信息也会由于过于宽松的相似度阈值被掩盖。因此,产生了新型序列聚类算法DADA2。DADA2软件把聚类结果称为ASVs (amplicon sequence variants),即简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,ASV能够分辨细菌序列单一核苷酸的差异。该流程2020年以后较为流行,ASVs序列准确性更高,但是缺点明显,即最终使用的有效Reads信息较少,会删除比较多的单序列。
Alpha多样性部分:
Alpha多样性:用于评估群落内生物种类的多少以及不同生物间相对丰度的数量指数。
Richness丰富度指数:一定空间范围内生物的丰富程度。
Pielou均匀度指数:群落中各个物种的相对密度。
Chao1、ACE:丰富度指数,估算样品中含OTU的数目,即所含物种的总数。
Simpson:反映的是优势种在群落中的地位和作用,数值越大,说明群落多样性越低。
Shannon:评估样品中物种组成的丰富度和均匀度,数值越大,说明群落多样性越高。
稀释曲线:从样本中随机抽取一定数量的序列,统计这些序列所代表的物种数目,并以序列数与物种数来构建曲线,用来说明样本的测序数据量是否能够完整的反应微生物群落。
PD:基于系统发育树来计算的一种多样性指数,它用各个样品中OTUs的代表序列构建出系统发育树的距离,将某一个样品中的所有代表序列的枝长加和,从而得到的数值。数值越大,群落多样性越高。
Alpha多样性相关公式:
Chao1
:是用chao1算法估计样本中所含OTU数目的指数,chao1在生态学中常用来估计物种总数,由Chao(1984)最早提出。公式如下:
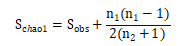
注:=估计的OUT数;=实际测到的OUT数;=只含有一条序列的OTU数目(如"singletons");=只含有两条序列的OTU数
ACE
:用来估计群落中OTU数目的指数,由Chao提出,是生态学中估计物种总数的常用指数之一,与Chao1的算法不同,其值越高代表群落物种种类越丰富。公式如下:
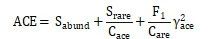
注:,丰富(丰度阈值大于n)物种数;,稀有(丰度阈值小于或等于n)物种数;仅包含1个个体的物种数;,稀有物种变异系数的估算值。
Shannon:用来估算样本中微生物多样性指数之一。它与Simpson多样性指数常用于反映alpha多样性。Shannon值越大,说明群落多样性越高。公式如下:
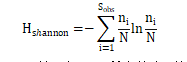
注:=实际观测到的OTU数;=第i个OTU所含的序列数;N =所有的序列数。
Simpson:用来估算样本中微生物多样性指数之一,由Edward Hugh Simpson(1949)提出,在生态学中 常用来定量描述一个区域的生物多样性。Simpson指数值越大,说明群落多样性越低。公式如下:
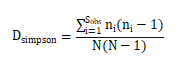
注:=实际观测到的OTU数;=第i个OTU所含的序列数;N =所有的序列数。
-
点赞 (0人)
- 收藏 (0人)

