
BLAST 比对结果的输出和结果解读
- 看不见的线
- 6585
- 2024-07-22 09:23:55
- 原创
BLAST比对结果通过outfmt参数指定输出格式,官方提供的输出格式是19种,值分别是0-18,以下是具体的介绍。
第 1种:outfmt 0
软件默认的输出格式,与网页版展示的比对结果类似。包括比对统计结果、比对序列等信息,示例如下:
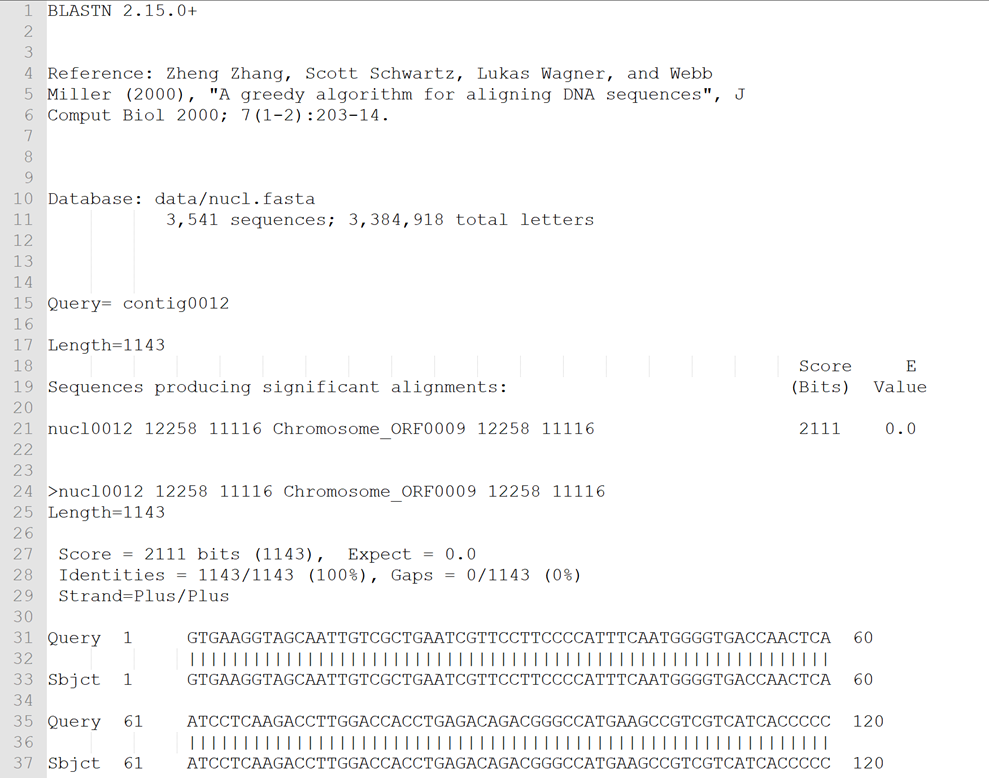
第 2种:outfmt 1
这种格式只保留了序列比对信息,给出输入序列,库中一致的用.表示,不一致的会显示不一致的碱基,可读性更强。
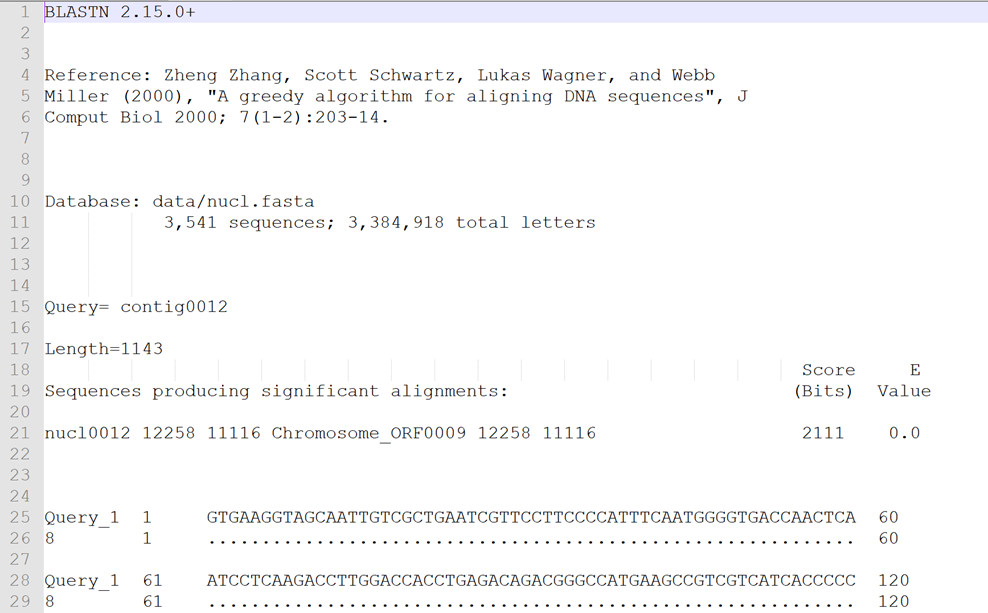
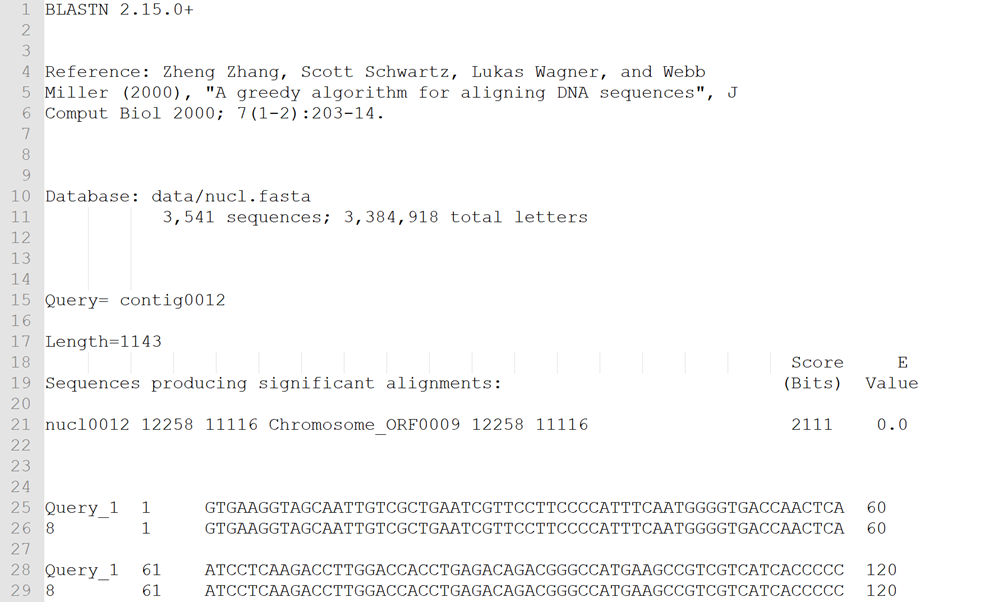
第 3种:outfmt 2
与第二种一致,只是序列展示有区别,所有比对的序列都保留碱基序列格式。
第 4-5种:outfmt 3/4
两种输出格式与第二、三种格式完全一致。
第 6种:outfmt 5
这种格式为xml格式的文件,很多种编程语言都可以直接解析。每一个输入序列及比对结果都会以嵌套格式进行整理。
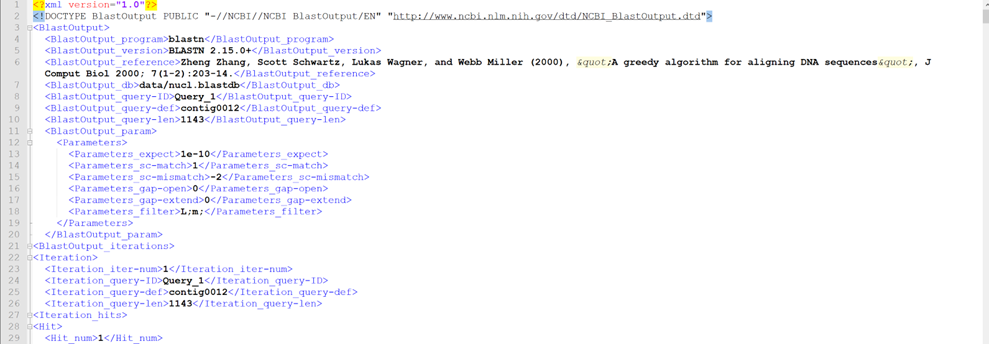
第 7种:outfmt 6
这种格式会输出比较多的统计信息,包括比对位置、得分、evalue等,可读性很强,一般都会选择输出这种格式。
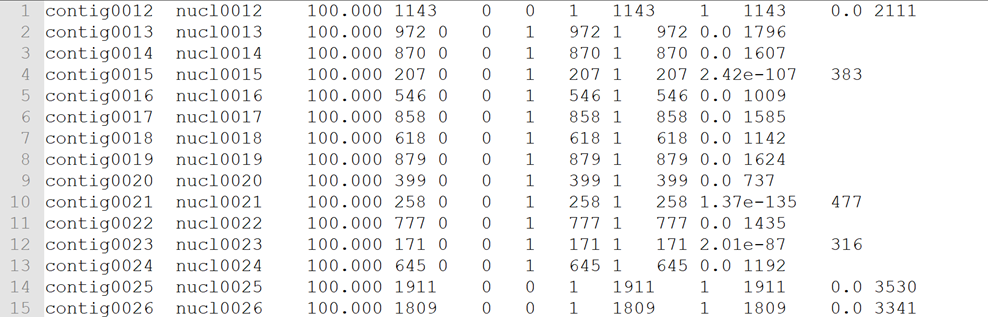
总共对应12列结果,每一列的含义如下:
query id:查询序列ID标识;
refer id:参考序列ID标识;
identity (%):序列比对的一致性百分比;
alignment length:符合比对的比对区域的长度;
mismatches:比对区域的错配数;
gap openings:比对区域的gap数目;
q.start:比对区域在查询序列上(query id)的起始位点;
q.end:比对区域在查询序列上(query id)的终止位点;
s.start:比对区域在参考序列上(refer id)的起始位点;
s.end:比对区域在参考序列上(refer id)的终止位点;
e-value:比对结果的期望值,将比对序列随机打乱重新组合,与数据库进行比对,如果功能越保守,则该值越低;
bit score:比对结果的bit score值。
第 8种:outfmt 7
这种格式基本与第7种一致,只不过每条序列加了说明信息,便于查看结果,相当于加了一个表头。

第 9-10种:outfmt 8/9
两种格式的编码方式是ASN.1,格式与xml格式类似,都是采用嵌套的方式进行展示。

第 11种:outfmt 10
这种格式的数据与第七、八种基本一致,只不过分隔符由tab键换成了逗号。

第 12种:outfmt 11
这种格式也是一种ASN.1格式的文件,只不过是用blast编码对数据进行了处理,序列信息使用一种叫NCBI2na的方法进行重新编码,具体可参照https://ncbi.github.io/cxx-toolkit/pages/ch_datamod。

其他格式 outfmt 12-18
多数使用json或者xml格式的文件进行编码,具体包括以下几个。
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
上述格式中,如果输入文件中存在多条序列,标注Multiple的会单独输出每条序列的比对结果。其中17输出sam格式的文件,18一般的比对时不能使用。
相关课程推荐:
课程名称:个人电脑上如何通过BLAST软件做比较基因组学分析
课程链接:https://college.mimazi.net/course/article-32.html
本节课的主要内容:
《个人电脑上如何通过BLAST软件做比较基因组学分析》
(1)BLAST软件可以解决的各类科学问题;
(2)BLAST软件在比较基因组方面的应用场景;
(3)实战:个人电脑上用BLAST+软件找同源基因。
-
点赞 (0人)
- 收藏 (0人)

