
使用Minimap2软件进行全长转录组测序数据比对
- 看不见的线
- 1784
- 2025-08-20 17:36:23
- 原创
Minimap2的安装
方法一:conda安装
#创建并进入conda环境
conda create -n Minimap2 -y
source activate Minimap2
#安装minimap2
conda install -c bioconda minimap2
方法二:编译安装
#获取并解压安装包
wget https://github.com/lh3/minimap2/archive/refs/tags/v2.28.tar.gz
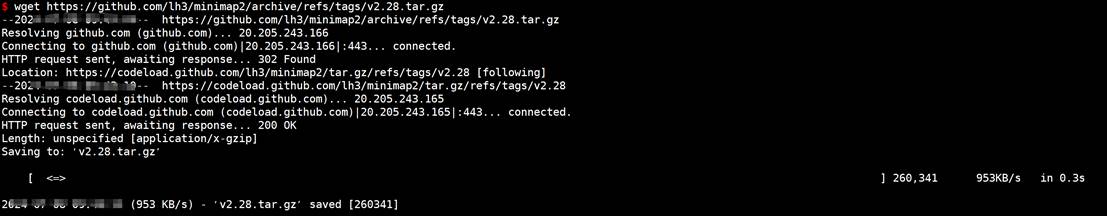
tar -zxvf v2.28.tar.gz
#切换到软件安装目录
cd minimap2-2.28
#运行安装
make
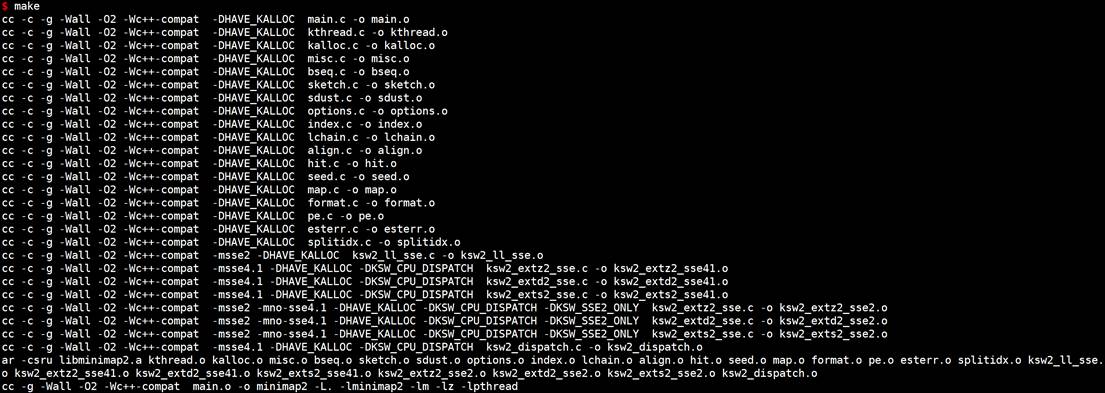
#添加环境变量,若不添加环境变量,可通过程序的绝对路径运行程序
# export PATH=软件安装路径:$PATH,此处为临时添加环境变量,也可将“export PATH=软件安装路径:$PATH”添加到 ~/.bashrc中,然后保存退出,使用 source ~/.bashrc命令让其生效
export PATH="$PWD:$PATH"
实战
1.准备参考基因组和转录组数据
确保已经有参考基因组序列(FASTA文件)和全长转录组测序数据(通常为FASTA或FASTQ格式)。
2.运行比对
使用以下命令进行全长转录组测序数据的比对。假设转录组数据文件名为 transcriptome.fq:
minimap2 -ax map-pb reference.fa transcriptome.fq > aln.sam # for PacBio CLR reads
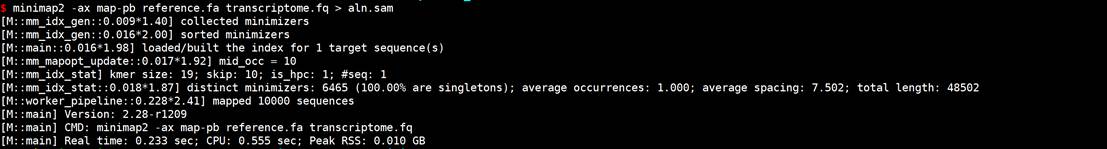
minimap2 -ax map-ont reference.fa transcriptome.fq > aln.sam # for Oxford Nanopore reads
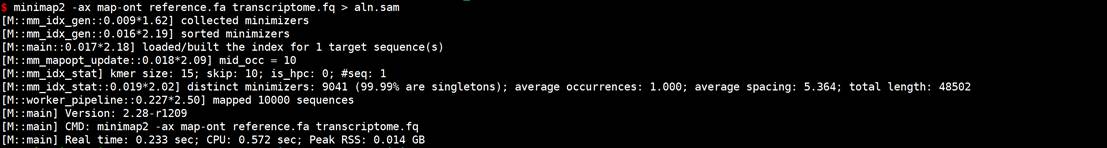
- 其中:
- -a:设置输出为sam格式
- -x:对不同类型数据,设置不同参数
- transcriptome.fq是转录组测序数据文件。
- aln.sam是输出文件名,将比对结果保存为SAM格式。
3.优化参数
根据数据特性和分析需求,可以调整一些参数以获得更好的比对效果。
例如:
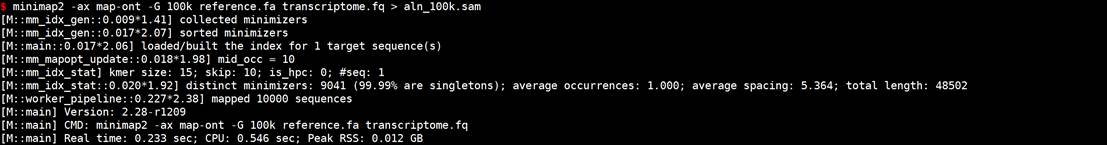
- -G参数控制最大内含子长度,默认是200k。根据需要调整,例如100k:
minimap2 -ax map-ont -G 100k reference.fa transcriptome.fq > aln_100k.sam
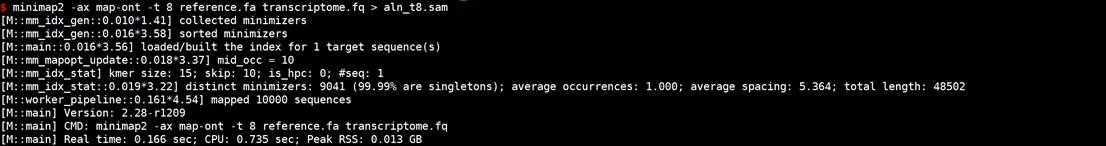
- -t参数设置线程数,可以加快比对速度。例如使用8线程:
minimap2 -ax map-ont -t 8 reference.fa transcriptome.fq > aln_t8.sam
密码子云平台
我们最近推出了密码子生信云平台服务(https://cloud.mimazi.net),包含免费细菌基因组云流程和各种生信分析小工具,无需安装软件、无需配置环境,即可一键化生成数据分析及可视化绘图结果,快来试试吧!
-
点赞 (0人)
- 收藏 (0人)

