
GFF和GTF文件的处理和应用
- 看不见的线
- 16491
- 2024-07-22 09:18:07
- 原创
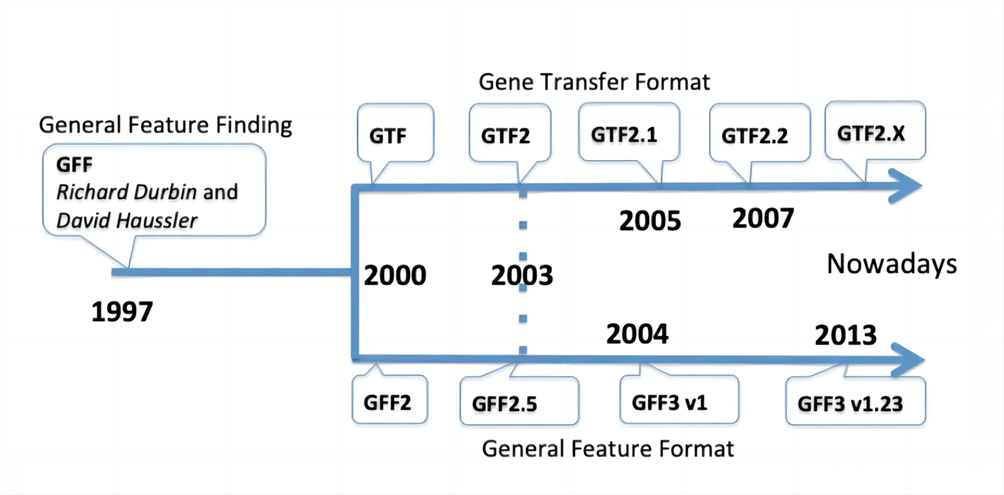
GFF和GTF文件的格式规则和应用场景
GFF(General Feature Format)和GTF(Gene Transfer Format)文件是用于描述基因组注释信息的制表符分隔的文本文件,在信息分析中一般需要从这两种文件中提取所需的注释信息。
GFF文件分为三个版本,其中GFF3是最新的标准,GTF文件实际上是GFF2的一个子集。这两种文件格式都包括了基因组特征的信息,例如基因、外显子、启动子等。
GFF全称为general feature format,这种格式主要是用来注释基因组。
GTF全称为gene transfer format,主要是用来对基因进行注释。
GFF格式规则:
- GFF3中,属性列是键值对形式,用分号分隔。
-包含九列,依次为:序列名、来源、特征、起始位置、终止位置、评分、链、相位和属性,详细描述如下:
seq_id:序列的编号。通常是染色体或者Contig ID,比如:Chr01或者 scaffold_1
source:注释的来源。一般会是预测用软件工具或者数据库
type:类型,此处的名词是相对自由的,建议使用符合SO惯例的名称(sequenceontology),如gene,repeat_region,exon,CDS等
start:上述type起始位点。从1起始计数
end:上述type结束位点
score:type的分数,可以是浮点数或者整数。与早期版本的格式一样,其语义定义不明确。建议将E-value用于序列相似性特征,将P-value用于从头基因预测。
strand:type位于参考序列的正链(+)或者负链(+),或者非链特征(.),对于链相关特征未知的也可以用问号(?)表示
phase:仅对注释类型为“CDS”的有效,表示起始编码的位置,有效值为0、1、2. (对于编码蛋白质的CDS,本列指定下一个密码子的起始位置。每3个核苷酸编码一个氨基酸,从0开始,CDS的长度,除以3,余数就是这个值,表示到达下一个密码子需要跳过的碱基数。“0” 表示密码子从该CDS的5‘端第一个核苷酸开始(即向前0个碱基),“1”相表示密码子从第二个核苷酸开始,“2”相表示密码子从第三个核苷酸开始。注意,GFF3 CDS的phase不应与类似概念frame混淆,frame的概念也是生物信息学中的常见概念。frame通常被计算为相对于完整开放阅读框(ORF)或密码子的开始的给定碱基值,而CDS phase表示相对于给定CDS特征的下一个密码子的开始。)
attributes:格式为tag=value的特征属性列表,不同的属性用分号分隔。一个键可以有多个值,不同值用“,”分割。注意如果描述中包括tab键以及“,=;”,要用URL转义规则进行转义。tag是区分大小写的,以大写字母开头的tag是预先定义好的,在后面可以被其他注释信息调用
| tag | 含义 |
| ID | 注释信息的编号,在一个GFF中必须唯一 |
| Name | 注释信息的名称,与ID不同,可以重复 |
| Alias | 特征的其他名称。例如基因的Sambol ID、locus ID等。与ID不同,文件中不要求唯一。 |
| Parent | 表示功能的上一级。Parent ID可用于将外显子指向转录本,将转录本指向基因等。特征可能有多个Parent。Parent只能用来表示关系层级的一部分 |
| Target | 核苷酸与核苷酸或蛋白质与核苷酸比对。该值的格式为target_id start-end[strand],其中strand是可选的,可以是“+”或“-”。如果target_id包含空格,则必须将其转义为 hex escape %20 |
| Gap | 比对结果的Gap信息,和Target一起,用于表明序列的比对结果 |
| Derives_from | 不同特征是时间关系而不是纯粹的结构关系时,用于消除一个特征与另一个特征之间的关系的歧义。多顺反子基因所必需的 |
| Note | 简单的文本信息 |
| Dbxref | 数据库交叉引用 |
| Ontology_term | 对 Ontology term的交叉引用 |
| Is_circular | 指示特征是否为环状 |
GTF格式规则:
-同样包含九列,格式与GFF相似,但属性列的键值对使用空格分隔,并以双引号括起来,详细信息如下:
seqname:序列名称。通常是染色体或者Contig ID,比如:Chr01或者 scaffold_1
source:注释的来源。一般会是预测用软件工具或者数据库
feature:注释信息类型(基因结构),这列信息是必须的,至少包括比如CDS、start_codon、stop_codon,还可以包含5UTR、3UTR、inter、inter_CNS、intron_CNS、exon、mRNA等等
start:上述feature起始位点。从1起始计数
end:上述feature结束位点
score:表示feature的存在和坐标的置信度,可以是一个浮点数或者整数,“.” 表示为空,就是不需要
strand:该feature位于参考序列的正链(+)或者负链(+)
frame:0表示阅读框的第一个完整密码子位于最5’端,1表示在第一个完整密码子之前有一个额外的碱基,2表示在第一整个密码子之前还有两个额外的碱基。注意,frame不是CDS长度除以3的余数(CDS mod 3)。如果链是“-”,则该区域的第一个碱基的值为end,因为对应的编码区域将在反向链上从end到start
attributes:应具有的格式为attribute_name“attribut_value”;,每个属性必须以分号结尾并且与下一个属性之间以空格分隔,并且属性的值应该用双引号包围。并且必须包含以下两个属性:
| attributes | 含义 |
| gene_id“value”; | 表示转录本在基因组上的基因座的唯一ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。 |
| transcript_id“value”; | 预测转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。 |
GTF与GFF比较:
GTF的第九列,通常为:
gene _ id "At1ge0001"; transcript _ id "At1g0ee01.1";
而 GFF的第九列,通常为:
ID =mrnae01; Name = abc ID =exon1; Parent =mrnae01ID =exon2; Parent =mrnae01
使用gffread软件可实现gtf和gff文件互转和序列提取:
gffread软件安装
方法一:conda安装
conda install bioconda::gffread -y
方法二:编译安装
# gffread软件的下载和编译
git clone https://github.com/gpertea/gffread
cd gffread
make release
#将gffread安装路径添加到环境变量(临时),需将此处路径替换为实际路径
export PATH="/mnt/Share/mawulin/class/class3/data/gffread:$PATH"
#查看帮助信息(检查安装是否成功)
gffread -h
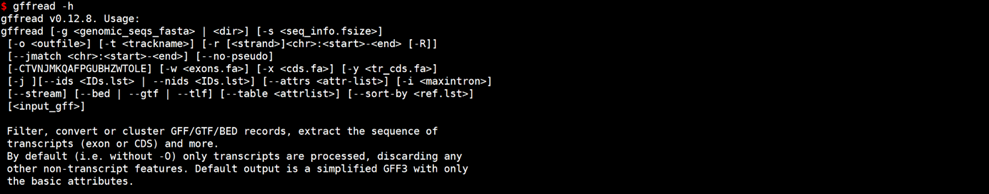
常用参数介绍:
-g 序列文件,即GFF/GTF文件第一列ID对应的序列文件。
-i 丢弃掉内含子大于的转录本(mRNA/transcript)
-r 起始和终止位置,填写示例100.10000即为输出与100到10000有重叠的所有转录组,也可以限制序列ID及链,填写示例:+Chr1:100..10000。
-R 丢弃掉此范围的转录本,与-r相反。
-U 丢弃掉 single-exon的转录本
-C 丢低调无CDS的转录本。
-V 丢弃掉含有移码突变的转录本。
-H 如果使用了-V,则重新检查并调整内含子相位,避免由于翻译起始位点选择的位置不对导致移码突变的产生。
-B 如果使用了-V, 对于单外显子基因,则重新检查相反的链,是否存在移码突变。
-N 丢弃掉多外显子基因剪接位点不是常见的 GT-AG, GC-AG or AT-AC序列。
-J 丢弃掉没有起始密码子或者终止密码子的转录本,仅保留有完整编码框的转录本。
--no-pseudo: 过滤掉含有 'pseudo'的注释信息
-M/--merge : 合并完全相同的或者存在包含关系的转录本。
-d:使用 -M 将合并信息输出到文件中
--cluster-only:类似于 --merge但是不合并转录本
-K 对于-M选项:also collapse shorter, fully contained transcripts with fewer introns than the container
-Q 对于-M选项:移除包含关系的转录本的限制条件:多外显子转录本将会合并,如果他们内含子位置完全一样,单外显子转录本只需要有80%一样即可合并。
--force-exons: 使GFF features的最小层级为exon
-E 对于重复的 ID或者 GFF/GTF其他潜在的格式问题给出警告信息。
-Z 将内含子小于4 bp的邻近的两个外显子合并为一个。
-w 输出每个转录本的外显子序列
-x 输出CDS序列
-W 对于 -w和 -x选项,输出外显子位置坐标到FASTA序列的ID中
-y 输出蛋白质序列
-L 将Ensembl GTF转换为 GFF3 conversion (implies -F; should be used with -m)
-o 输出"filtered"后的GFF文件。
-T -o参数将输出 GTF格式。
#查看示例数据
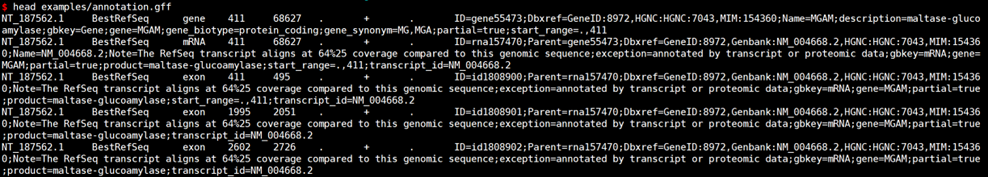
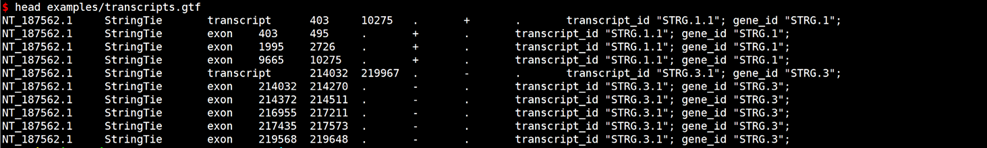
# GFF转换GTF
gffread examples/annotation.gff -T -o out.gtf
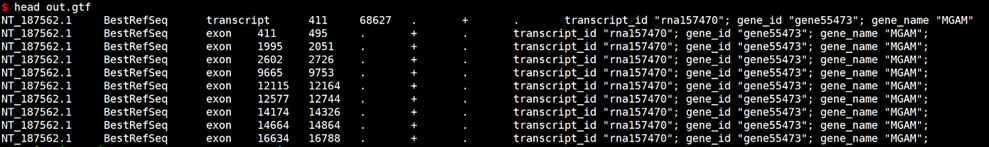
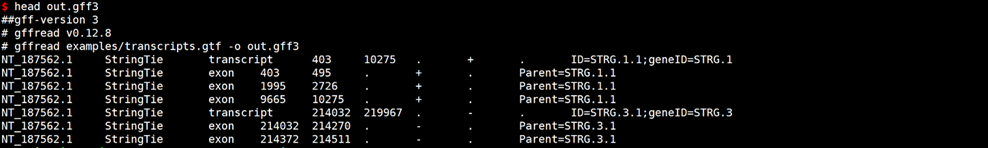
# GTF转换GFF3
gffread examples/transcripts.gtf -o out.gff3
#根据GFF或者GTF提取CDS,蛋白质和外显子序列
gffread examples/annotation.gff -g examples/genome.fa -x cds.fa -y pep.fa -w cdna.fa


推荐课程
【课程】微生物比较基因组精品系列课——全套自学必入的系统课程
课程链接:微生物比较基因组精品系列课【全套】
【课程】铜绿假单胞菌基因组研究和分子分型实战
课程链接:铜绿假单胞菌基因组研究和分子分型实战
【课程】微生物比较基因组与群体进化——基因组变异专题研究
课程链接:微生物比较基因组与群体进化
【课程】微生物分子分型-MLST课程——分型全套(含理论、软件、方法)
课程链接:微生物分子分型-MLST课程
【课程】基因组结构分析神器Easyfig实操精品课
【课程】BRIG绘图——结构比较专题2
【课程】肺炎克雷伯菌基因组学研究综合指南
课程链接:肺炎克雷伯菌基因组学研究综合指南
【课程】微生物基因组生信必学课程
课程链接:微生物基因组生信分析必学课程
【课程】微生物生防菌研究
课程链接:生防菌的系统化研究
专题材料
【资料】耐药专题材料
【资料】生防专题材料
请添加唯那生物技术客服小唯的微信号winnerbio01,备注“耐药专题”或“生防专题”,立马获取。
更多专题推荐
-
点赞 (0人)
- 收藏 (0人)
-
上一篇: 没有了
- 下一篇: MLST、rMLST、wgMLST、cgMLST介绍

