
将FastA、FastQ文件拆分成多个文件
- 看不见的线
- 21
- 2025-06-25 13:41:07
- 原创
Seqkit2是 Seqkit的新版本,是一个专门用于处理和分析生物序列数据的软件工具。它支持多种序列数据格式,包括FASTA、FASTQ等,并提供了一系列有用的功能,如数据处理、过滤、统计、格式转换等,是生物信息学领域中常用的工具之一。以下是使用seqkit实现将FastA、FastQ文件拆分成多个文件。
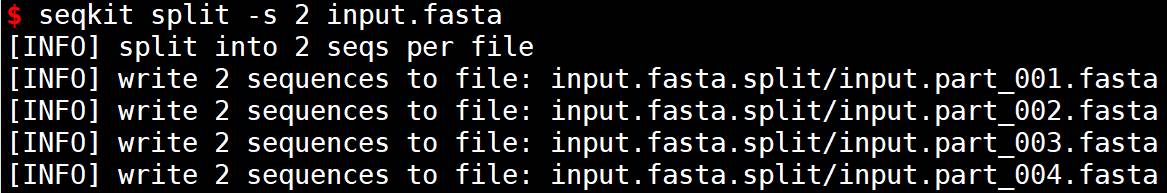
- 1、基于指定序列数拆分文件(拆分后的文件将按照原始文件名加上后缀 _1, _2, _3等进行命名)
# -s 2指定了每个输出文件包含的最大序列数为1000
# input.fasta是要拆分的输入FASTA文件
seqkit split -s 2 input.fasta
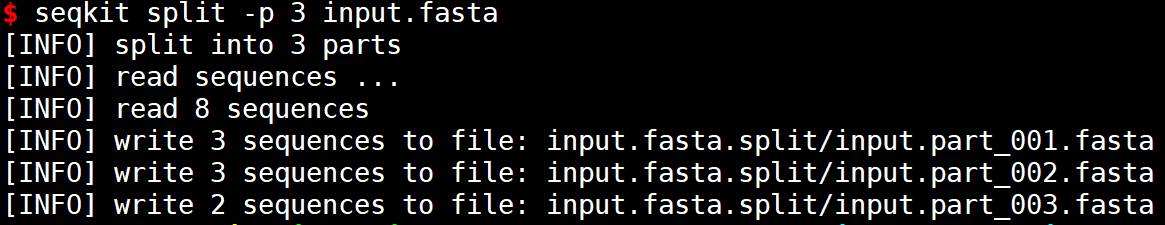
- 2、将文件拆成特定份数(拆分后的文件将按照原始文件名加上后缀 _1, _2, _3等进行命名)
# -p 3指定了要将输入文件拆分为3个文件
# input.fasta是要拆分的输入FASTA文件
seqkit split -p 3 input.fasta
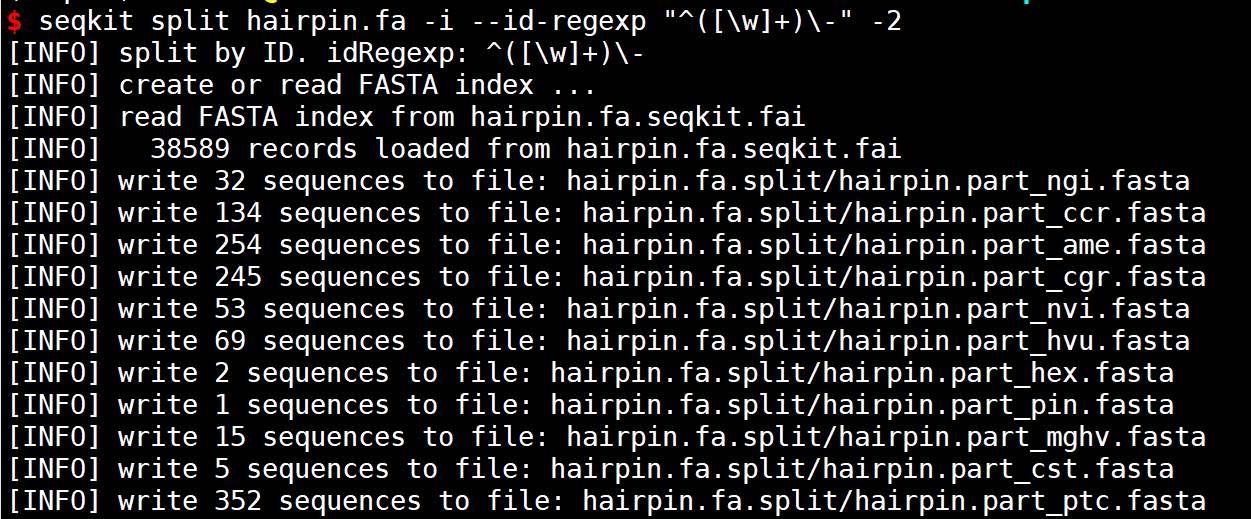
- 3、基于ID拆分文件
# --id-regexp "^([\w]+)\-":使用正则表达式提取 ID的一部分。在这里,正则表达式 ^([\w]+)\-匹配 ID开头的字母数字字符([\w]+),直到遇到第一个 -符号
# -2:启用两遍模式,以降低内存使用量,特别适用于大文件
seqkit split hairpin.fa -i --id-regexp "^([\w]+)\-" -2
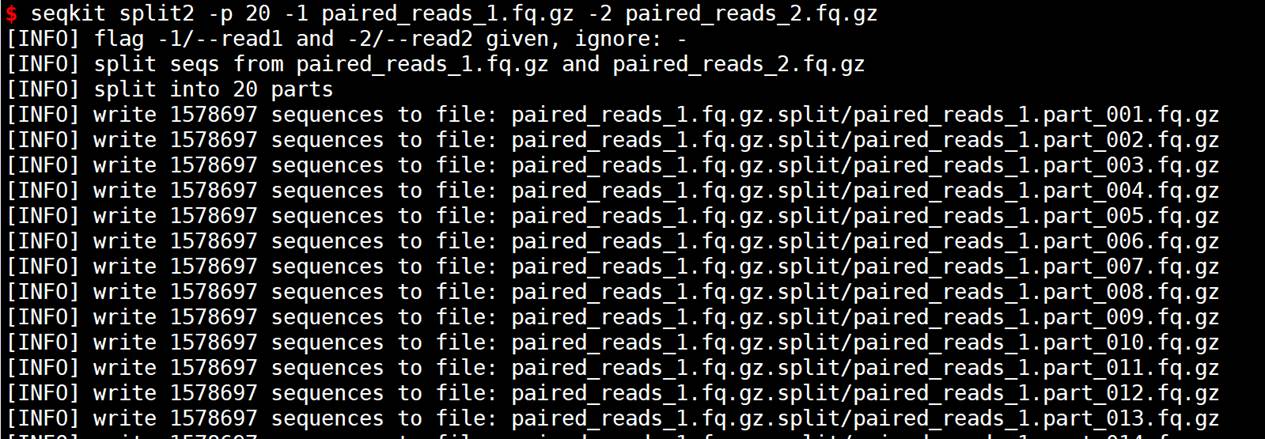
- 4、同时拆分双端测序fq文件
# -p 20指定了同时拆分为20个文件,因为双端测序通常包含两个文件(每个文件对应一个端)
# -1 paired_reads_1.fq.gz -2 paired_reads_2.fq.gz是要同时拆分的输入双端测序FASTQ文件
seqkit split2 -p 20 -1 paired_reads_1.fq.gz -2 paired_reads_2.fq.gz
-
点赞 (0人)
- 收藏 (0人)
-
上一篇: 没有了
- 下一篇: Easyfig罕见问题1

