
Bowtie2软件的安装和使用
- 看不见的线
- 71
- 2025-07-25 11:28:18
- 原创
1. Bowtie2软件的适用范围和应用场景
Bowtie2是将测序后的 reads与长参考组的比对工具 (适用于将长度大约为50~1000bp的reads与相对较长的基因组,如哺乳动物,进行比对)。
Bowtie2使用FM索引(基于Burrows-Wheeler Transform或 BWT)对基因组进行索引,以此来保持其占用较小内存。对于人类基因组来说,内存占用在3.2G左右。Bowtie2支持间隔,局部和双端对齐模式。可以同时使用多个处理器来极大的提升比对速度。
通常是比较基因组学(包括 variation calling,ChIP-seq,RNA-seq,BS-seq)管道的第一步。可以处理非常长的 Reads(即10~100kb),但它针对近期测序仪产生的 Reads长度和误差模式进行了优化,如Illumina HiSeq 2000,Roche 454和Ion Torrent仪器。
2. Bowtie2软件的下载与安装
方法一:使用源码安装
#下载安装包
#解压安装包
unzip bowtie2-2.5.4-sra-linux-x86_64.zip
#跳转到解压后目录,并获取其路径
cd bowtie2-2.5.4-sra-linux-x86_64 && pwd
#将前一步获取的路径追加到 ~/.bashrc文件的末尾
echo PATH=$PATH:/path/to/software/bowtie/bowtie2-2.5.4-sra-linux-x86_64 >> ~/.bashrc
#重新加载 ~/.bashrc文件
source ~/.bashrc
#验证文件是否正确添加(若输出的是 bowtie2的路径,则表示设置成功)
which bowtie2

方法二:使用conda安装
conda install bowtie2 -y
3. Bowtie2软件的参数解读和使用建议
3.1参数解读
#查看帮助信息
bowtie2 -h
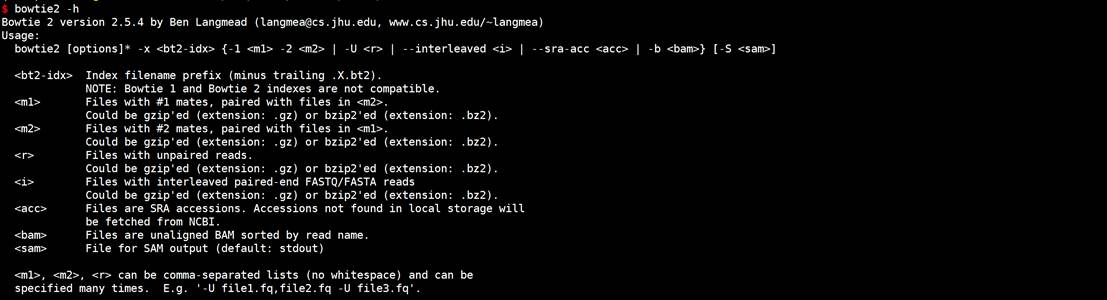
3.2 bowtie2使用方法
#查看参考序列(此处以bowtie2自带示例数据为例)
head lambda_virus.fa

#使用参考序列构建索引,index为索引前缀
bowtie2-build lambda_virus.fa index


#目标序列与参考数据库进行比对

#对双端序列比对
bowtie2 -x index -1 reads_1.fq -2 reads_2.fq -p 32 -S example_pair.sam


#对单端序列比对
bowtie2 -x index -U reads_1.fq -p 32 -S example_single.sam


#使用samtools将sam文件转换为bam文件以供后续分析使用
#需提前安装和配置samtools
samtools sort example_pair.sam > example_pair.bam
samtools sort example_single.sam > example_single.bam
-
点赞 (0人)
- 收藏 (0人)
-
上一篇: 没有了
- 下一篇: Easyfig罕见问题1

