
BWA软件的安装和使用
- 看不见的线
- 1910
- 2025-07-24 09:26:00
- 原创
1. BWA软件的适用范围和应用场景
bwa是一款将序列比对到参考基因组上的软件,包含了以下3种算法:
BWA-backtrack:用于进行Illumina reads的比对,reads的长度最大为100bp。
BWA-SW:用于比对long-read,支持的长度为70bp-1Mb;同时支持剪接性比对(split alignments)。
BWA-MEM:最新的算法。和BWA-SW的适用性一致,但是更加快速和准确;同时与BWA-backtrack相比,在对70bp-100bp reads的比对上有更优的性能。通常情况下选择BWA-MEM算法就好。
2. BWA软件的下载与安装
可以直接去github上下载(https://github.com/lh3/bwa),也可以使用git clone直接拷贝到本地,如:
git clone https://github.com/lh3/bwa.git
进入bwa目录,直接make,等待安装完成即可。
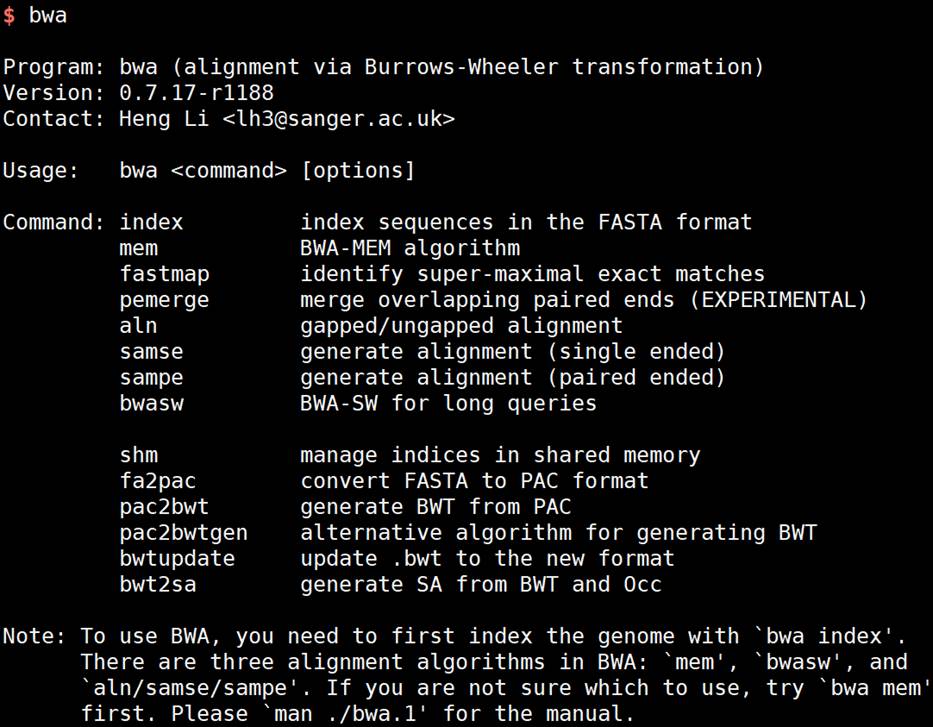
安装完成后,输入bwa,就能查看帮助信息。
3.实战:使用BWA软件生成质粒基因组的SAM文件
使用BWA整个比对过程主要分为两步,第一步建索引,第二步进行比对。
示例数据:
├── plasmid_genome.fasta………………………………………………质粒基因组序列
├── plasmid_R1.fq.gz……………………………………………………测序数据read1
├── plasmid_R2.fq.gz……………………………………………………测序数据read2
操作:
$ bwa index plasmid_genome.fasta -p plasmid_index……………………建立索引
$bwa mem plasmid_index -t 16 plasmid_R1.fq.gz plasmid_R2.fq.gz > plasmid_pe.sam……使用bwa-mem算法进行双端比对
结果:
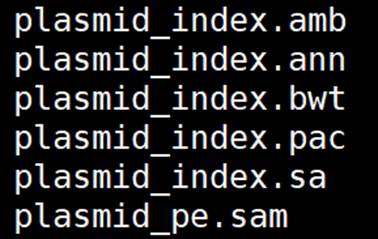
-
点赞 (0人)
- 收藏 (0人)

